Qumica Biolgica Patolgica GENETICA y TECNICAS DE BIOLOGIA
")
Química Biológica Patológica GENETICA y TECNICAS DE BIOLOGIA MOLECULAR I- Parte Tema: 1 (1) Dra. Silvia Varas svaras@unsl. edu. ar

Química Biológica Patológica Presentación del CURSO Integrantes: Dra. Maria Sofía Giménez Dra. Silvia Mabel Varas Lic. María Rosa Fernández Dra. Mariana Lucila Ferramola Dra. María Gabriela Lacoste Lic. José Luis Arias

El presente curso: n Es un curso en permanente modificación y actualización de sus contenidos n Precisa conocimiento previos n Integra conocimientos obtenidos en cursos previos de la carrera.

Herramientas de comunicación: BLOG: http: //qbpatologica. wordpress. com n Cartelera 2º Piso Barco n Horarios de Consulta: Viernes 15 hs n

LIBRO del Curso Bioquímica Molecular Gimenez MS y col. n Capítulos: n Cáncer, pag. 128 n Apoptosis, pag. 108 n Fibrosis Cística, pag. 253 n Porfirias, pag. 293 n Fenilcetonuria, pag. 354 n


Actividades Extras y Obligatorias: Tema 5. Gota n Tema 10: Glucogenosis n Guía de estudio n

RESUMEN n En la cursada se va ver y examinar ≈60% del programa
")
Química Biológica Patológica GENETICA y TECNICAS DE BIOLOGIA MOLECULAR I- Parte Tema: 1 (1) Dra. Silvia Varas svarasl@unsl. edu. ar

Ruta o Vía Metabólica A B C D E F G P

Ruta o Vía Metabólica

Una ruta metabólica se realiza en alguna organela en particular

 “Trastorno bioquímico determinado genéticamente en el que la falta")
Errores congénitos del metabolismo (ECM) “Trastorno bioquímico determinado genéticamente en el que la falta (o hay una alteración) de una proteína que origina un bloqueo metabólico consecuencias clínicas”

Antecedentes Garrod 1908: albinismo, alcaptonuria, cistinuria, pentosuria familiar n Følling 1934: descripción de la fenilcetonuria (PKU) n Métodos cromatográficos 1940 -1980 n ADN - Bases moleculares de la genética 1950 -1960 n Genoma humano 2001 -2013 Genómica n Productos génicos 1990 -2013 Proteómica n
")
n Garrod, 1903 The Metabolic and Molecular Bases of Inherited Disease (MMBID)

Alteraciones primarias de los ECM A 1 extracelular Membrana A 2 B intracelular C 4 3 D Apoenzima + cofactor 1. Alteración del transporte de membrana. 2. Bloqueo enzimático de B a C: a) Deficiencia de C. b) Acúmulo de A y B (soluble ó insoluble). 3. Aumento de D (vía alternativa) por exceso de B. 4. Defecto entre la interacción de apoenzima-cofactor obligatorio.

Defectos I: Receptores de membrana: - RLDL, LRP 1, LRP 2, etc. - Hormonas: TSH, insulina, glucagón n n - Transportadores de MEMBRANA PLASMÁTICA: Familia ABC( A 1, C 2, C 7) Familia SLC (SLC 3, SLC 7 aa) Simporter NIS Pendrina

Defectos II: n - Enzimas: Glu 6 P-D n Cofactores: GALT - Apo CII UGT-1 A 1 Tiroperoxidasa - Biotina ALA dehidrasa PBG deaminasa LPL n - Proteínas: globina Tiroglobulina Distrofina

Tipos n n Aminoácidos Lípidos y lipoproteínas n n n Proteínas n n Lipidosis, mucolipidosis Dislipemias Glucogenosis Galactosemia, fructosemia Malabsorción Hb Porfirinas Purinas, hiperuricemias Hormonas n n Tiroides Suprarrenal Enfermedades lisosomales n Hidratos de carbono n n n Enfermedades del sistema de transporte a traves de membrana n n Fibrosis Cistica Trastornos Tejido muscular n n Esfingolipidosis Mupolisacaridosis Lipidosis Otros Distrofias Enfermedades Multifactoriales n n Diabetes Obesidad

Epidemiología Enfermedad Incidencia PKU, FENILCETONURIA 1/10. 000 Hipotiroidismo 1/2. 500 Galactosemia 1/10. 000 Deficiencia de Biotidinasa 1/70. 000 Hipoplasia suprarrenal congénita 1/12. 000 Fibrosis quística 1/2. 500 Deficiencia de la Biotinidasa 1/70. 000 Distrofia muscular de Duchenne 1/4000 Hemoglobinopatias (talasemias) 1/500 Drepanocitosis (raza negra) 1/1. 400

Genetica mendeliana Pisum sativum Guisante, arveja o chícharo Fraile Gregor Johann Mendel 20 -7 -1826/ 6 -1 -1884 • 1843, en el convento de agustinos de Brünn

Desordenes Genéticos: Categorías 1. 2. 3. Cromosomales Monogenéticos o Mendelianos Multifactoriales n n 7/1000: Dominantes 2, 5/1000: Recesivos 0, 4/1000: Ligado X Mitocondrial

HERENCIA AUTOSOMICA q q q Recesiva La herencia afecta, habitualmente, a las uniones de dos heterocigotas Riesgo de un 25% enfermo de padres heterocigotas. Dos individuos afectados tienen 100 % de descendencia afectada. Suele existir alto grado de consaguinidad. Ex: Albinismo, Fibrosis cistica, Talasemia, etc. q q Dominante Todos los individuos afectados deben tener al menos un padre afectado. Una persona afectada tiene un 50% de posibilidades de trasmitir el rasgo a c/u de sus hijos. Dos individuos afectados pueden tener hijos no afectados. En algunos casos el fenotipo homocigoto puede ser más grave que el fenotipo heterocigoto o ser diferente a aquél.

Herencia ligada a X Dominante v v Todas las hijas de los varones afectados están afectadas y ninguno de los hijos está afectado. Una mujer heterocigota afectada trasmitirá el rasgo a la mitad de sus hijos, estando igualmente afectados los varones y mujeres. En promedio habrá doble n° de mujeres afectadas que de varones. Ex. Deficiencia de Glu 6 P deshidrogenasa, distrofia muscular, etc. Recesiva v v Están afectados los varones hemicigotos y las mujeres homocigotas Los varones afectados trasmiten el gen a todas sus hijas, pero a ninguno de sus hijos. Las hijas de varones afectados son habitualmente heterocigotas (portadoras) y no estan afectadas. Ex: daltonismo o ceguera para los colores verde y rojo.
 Regiones Bandas 3 P 1 q 1. 2")
Nomenclatura de los Cromosomas (Francia, 1971) Regiones Bandas 3 P 1 q 1. 2 2 1 Banda Q Región Brazo P (Petit) Q( Queve) Cromosoma 1

Patogenia n Alteraciones del DNA nuclear o mitocondrial n n Mutaciones Alteraciones de la síntesis/procesamiento del RNA Genes AB BC CD DE Proteínas (Enzimas) ab bc cd de A B C D Metabolismo intermediario E
 cerca del 11% de las sustituciones en regiones")
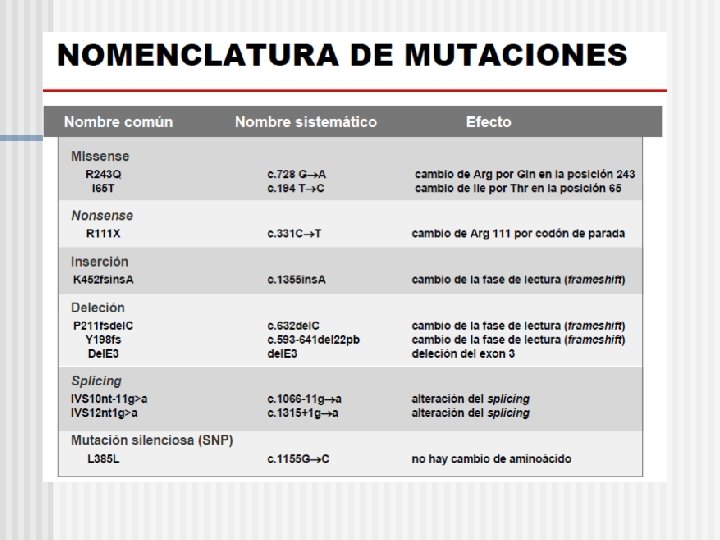
Mutación Puntuales ØMutaciones sin sentido (nonsense) cerca del 11% de las sustituciones en regiones codificantes. Es el cambio de una base por otra que genera un codón stop. ØMutaciones con cambio de sentido (missense) cerca del 45% de las mutaciones. Existe un cambio de base genera otro codón para un aa distinto al original. ØMutaciones silenciosa, el cambio de una base no marca un cambio de los aa. ØMutaciones de elementos de control o regulatorias, (1, 8%). ØMutaciones en secuencias consenso del splicing (9, 6%).
 ØPequeñas deleciones (15, 8) ØRearreglos Complejos (0, 9%)")
Mutaciones Del/Ins ØGrandes deleciones (6, 1%) ØPequeñas deleciones (15, 8) ØRearreglos Complejos (0, 9%) ØGrandes Inserciones y Duplicaciones (1, 2%) ØExpansión de repeticiones de trinucleótidos (0, 3%)
 6,")
Espectro de diferentes tipos de mutaciones en genes humanos (Human Gene Mutation Database) 6, 1% 15, 8% 9, 6% 11, 2% Mutaciones sin sentido Mutaciones con cambio de sentido 45, 1% n 14. 363 mutaciones en 783 genes

Resumen de Mutaciones informadas

Mutaciones Puntuales: Sustituciones de una única par de base en genes que causan enfermedades hereditarias

DELECIONES DE GENES n n n Las deleciones son responsables de mas 500 enfermedades hereditarias en humanos. Y estas deleciones pueden ser clasificadas en base a la longitud del ADN delecionado. Algunas deleciones consiste de solo unas pocas pares de bases hasta varias cientos de kilobases

GRANDES DELECIONES DE GENES n Grandes deleciones son comunes en la distrofia muscular de Duchenne, GH, RLDL y el gen de 1 -globina, SAG: 21 hidroxilasa, etc.

15, 8% PEQUEÑAS DELECIONES n Existe un total de 2. 368 deleciones de genes causantes de enfermedades con una longitud de 20 bp o menos

MUTACIONES PUNTUALES QUE AFECTAN SPLICING R: A o G Y: C o T - La porcion 5’ se llama “sitio dador” y la 3’ “sitio aceptor”. - Todos los genes eucariotas tienen una secuencia GT en el extremo 5’ y una secuencia AG en el extremo 3’ de cada intron. - Hay un punto de ramificación cercano al sitio 3’ del intron ( es un sitio consenso muy conservado CTRAY

NOMENCLATURA DE MUTACIONES DE SPLICING http: //www. hgvs. org/mutnomen/recs-DNA. html

9, 6% 58% Total 8% Creación nuevos sitios 34%

TIPO DE MUTACION DEFINE LA TECNICA DE BIOLOGIA MOLECULAR A USAR PARA EL DIAGNOSTICO MOLECULAR
- Slides: 43