Qumica Biolgica Patolgica GALACTOSEMIA 2017 Tema 9 Bolilla
 Dra. Silvia Varas svaras@unsl. edu.")
Química Biológica Patológica GALACTOSEMIA 2017 Tema: 9 (Bolilla 9) Dra. Silvia Varas svaras@unsl. edu. ar

TEMA 9: n n Galactosemia. Manifestaciones clínicas. Metabolismo de la galactosa en la galactosemia. Defectos enzimáticos. Metabolito tóxico. Diagnóstico. Prevención. Detección de portadores. Fructosuria. Metabolismo de la fructosa. Fructosuria esencial. Intolerancia hereditaria a la fructosa. Cuadro clínico. Mecanismo bioquímico. Diagnóstico.

Origen de Galactosa Leche n Ligada con enlaces tipo en ciertas legumbres, verduras y frutas. n En forma de galactocerebrósidos y gangliósidos en algunas vísceras de animales n

80% Vía Glicolítica 20% GPr y GLp Glucosa-6 -Fosfatasa GLUCEMIA SANGRE Via Glicolitica Piruvato Acetil Co. A Ciclo Krebs CO 2+ H 2 O Glucosa-6 - Deshidrogenasa Via de las Pentosas D-Ribosa-5 -P +NADPH + CO 2

Vías de Metabolización de la Galactosa

Vías de Metabolización de la Galactosa
, Premio Nóbel, 1970")
(1906– 1987), Premio Nóbel, 1970

n “por su descubrimiento de los nucleótidos de azúcar y su rol en la biosíntesis de carbohidratos” Papel de los UDPazucares: en los mamíferos, la mayoría de los grupos glucosilos son donados por UDP-azúcares

Metabolismo Normal Galactosa: Vía Leloir

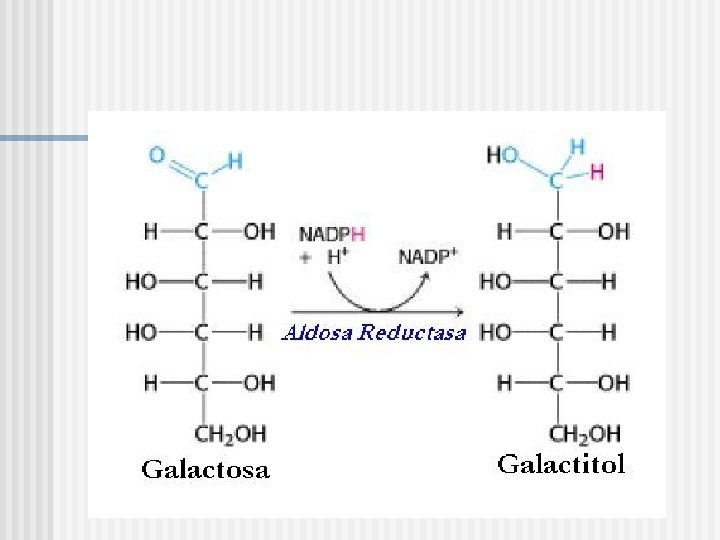
VIAS ALTERNATIVAS DE METABOLIZACION DE GALACTOSA Reducción a galactitol Oxidación a galactonato La “vía de la pirofosforilasa” y bypass de GALT

Desarrollo de cataratas “vía de la pirofosforilasa” Vía Glicolítica galactitol Glu-1 -P + UTP Aldosa Reductasa (AR) ADP ATP GALK Dieta -D-galactosa (1) IMPasa GALT Gal-1 -P UDP-Glu (4) (3) UGP (Pirofosforilasa) UDP-Gal GALE PPi (2) Galactosa Deshidrogenasa (GDH) UGP (Pirofosforilasa) UTP galactonato Vía de las Pentosas Acido D-Galactónico Acido -ceto-D-Galactónico D-Xilulosa Fosfato Via de las Pentosas Fosfato CO 2 UTP-glucosa/galactosa pirofosforilasa IMPasa: Inositol Mono Fosfatasa

Galactosa es un monosacárido esencial? PRODUCCION ENDOGENA DE GALACTOSA ANTE UNA DIETA LIBRE DE GAL UTP-glucosa/galactosa pirofosforilasa” Inositol monofosfatasa (IMPasa)
 UGP (UTP-glucosa/galactosa Pirofosforilasa) ADP ATP GALK Dieta")
Vía Glicolítica Producción Endógena de Galactosa (2) UGP (UTP-glucosa/galactosa Pirofosforilasa) ADP ATP GALK Dieta UTP + Gal-1 -P Glu-1 -P -D-galactosa GALT UDP-Gal (3) IMPasa UDP-Glu (1) GALE
 (Tipo II) Galactosa 1 - Fosfato Uridil")
DESORDENES HEREDITARIOS DEL METABOLISMO GALACTOSA Galactoquinasa (GALK) (Tipo II) Galactosa 1 - Fosfato Uridil Transferasa (GALT) (Tipo I) Uridina difosfato galactosa 4 - Epimerasa (GALE) (Tipo III)

Galactosemia Clásica David J. Timson: The molecular basis of galactosemia — Past, present and future. Gene 589 (2016) 133– 141
 El gen GALK 1 se encuentra en el brazo largo (q) del")
Galactoquinasa (GALK) El gen GALK 1 se encuentra en el brazo largo (q) del cromosoma 17 en la posición 24 n La enzima se expresa en hígado, GR, leucocitos, fibroblastos, placenta y varios tejidos fetales y adultos en humanos n El dominio N terminal de la enzima monomerica se muestra en azul y el dominio C-terminal en rojo. Se muestra el sitio activo donde se une galactosa y fosfato inorgánico
 n n Se expresa en hígado, ID,")
Galactosa 1 - Fosfato Uridil Transferasa (GALT) n n Se expresa en hígado, ID, ovarios, cerebro, fibroblastos y eritrocitos n El gen que codifica GALT esta localizado en el cromosoma 9 p 13 y tiene una extensión de 4, 3 Kb, constituido por 11 exones. Las dos subunidades del dímero de la enzima se muestran en color rojo y azul. Se indica la posición de los metales en formas de esferas. En el sitio activo de la enzima se encuentra unido UDPglucosa. Tiene regulación dietaria y hormonal

Gen GALT 3’ 5’ Exón: 1 Tamaño 82 2 3 4 5 170 76 49 130 6 7 57 123 (nt) 4, 3 Kb Proteína: 379 aminoácidos 8 9 133 83 10 11 155 78
 Se expresa en hígado, ID, fibroblastos y eritrocitos")
Uridina difosfato galactosa 4 Epimerasa (GALE) Se expresa en hígado, ID, fibroblastos y eritrocitos n El gen GALE mapea en el cromosoma 1 p 36. El gen incluye 12 exones que se extienden alrededor de 4 kb de ADN genómico n Cada subunidad de la enzima dimérica se plega en dos dominios. Los dominios N y C terminal están en color azul y rojo, respectivamente. Unido al sitio activo NADH y UDP-glucosa.

Biología Molecular Galactoquinasa Al menos 25 mutaciones diferentes han sido identificados en los lugares de GALK galactoquinasa en pacientes. n Frecuencia: 1/214. 470 n La enzima humana incluye 392 aminoácidos con una masa molecular de 42 k. Da n Algunas de las mutaciones son: 1569 C T en el exón 2 (R 68 C), 7093 C T en el exón 6 (T 288 M), 7538 G C en el exón 8 (A 384 P) y una deleción de un par de bases en el exón 5 (2833 del C).

Galactosa-1 -Fosfato Uridil Transferasa Las 6 mutaciones más comunes p. Q 188 R, p. S 135 L, p. K 285 N, p. L 195 P, p. T 138 M y p. Y 209 C son el 80% de los casos de galactosemia clásica. Una 7ª mutación IVS 2 -2 A G ha sido descrita en la población hispana. Mutación N 314 D: Variantes
 Frecuencia: Galactosemia Clásica: 1: 42. 894 Deficiencias Parciales: 1:")
Galactosa-1 -Fosfato Uridil Transferasa (GALT) Frecuencia: Galactosemia Clásica: 1: 42. 894 Deficiencias Parciales: 1: 14. 623

VARIANTES ALELICAS DE GALT

La variante Duarte es un alelo polimorfico del gen GALT que resulta en actividad alterada de la enzima. Este polimorfismo común en la población es sin signos clínicos o síntomas

n n Deficiencia de GALT: Alelos Duarte: La mutación N 314 D esta asociada con dos variantes de galactosemia que tienen actividad de GALT alterada pero con fenotipos subclínicos diferentes. Duarte-1 (Los Angeles) n Duarte-2 (D 2) Esta asociado con n Esta asociado con aumento de la actividad disminución de la actividad de GALT (110 -130% de de GALT lo normal) n El polimorfismo N 314 D esta en un un ligamiento en desequilibrio de desequilibrio con 3 ligamiento con la sustituciones intrónicas (IVS 4 nt-27 g c; IVS 5 ntsustitución silenciosa L 218 L (c. 652 C T). 24 g a y IVS 5 nt+62 g a) y una deleción de 4 pb en el promotor de GALT n La frecuencia de los alelos La frecuencia de los D 2 es de 5, 1 alelos D 1 es de 2, 7

Promotor gen GALT El promotor GALT presenta un sitio AP-1 desde − 126 a − 119 bp (TCAG) y dos motivos E-box desde − 146 a − 141 bp (CAGGTG) y desde − 117 a − 112 bp (CACGTG). La deleción − 119 a − 116 GTCA remueve los dos primeros nt del motivo E-box.

Para la genotipificación: Se consideran como genotipo D 2 a los individuos con al menos una copia de N 314 D y una copia de la deleción de 4 pb y n Presenta el genotipo D 1 al individuo que presente al menos una copia de N 314 D y ausencia de la deleción de 4 pb n

Relación Genotipo/Fenotipo n n El Alelo Duarte puede tener significancia clínica con otras mutaciones en el gen GALT, llamadas galactosemia DG (genotipo Duarte/Galactosemia). La combinación de una mutación clásica (G) y la variante D muestra una disminución de la actividad de GALT pero no resulta típicamente en los síntomas de galactosemia clásica

Tabla II: Diagnostico de sospecha de la Galactosemia I-Clínica de sospecha II- Bioquímica Inespecífica A- Síntomas Tóxicos A- Disfunción Hepática Vómitos Rechazo del alimento Depresión neurológica Hiperbilirrubinemia Hipoalbuminemia Déficit complejo protrombina GOT, GPT, GGT, LDH Acidos Biliares plasmáticos. Hipoglucemia B- Afectación oftalmológica. Cataratas C- Fracaso hepático grave Ictericia Hepato-esplenomegalia Ascitis Diátesis hemorrágica D- Tubulopatía proximal B- Tubulopatía proximal renal Acidosis hiperclorémica Glucosuria Aminoaciduria Albuminuria III- Bioquímica Específica A-Galactosuria B- Aumento de galactosa en plasma C- Aumento de Gal-1 -P en eritrocitos D- Galactitol en plasma y orina

Diagnostico Bioquímico n. Determinación de metabolitos n. Determinación de actividad enzimática

Determinación de metabolitos y actividad enzimática: n Gal-1 -P en eritrocitos n Gal TOTAL en eritrocitos Resumen de los niveles de metabolitos Deficiencia Galactosa (Plasma u Orina) Gal-1 -P (sangre) GALK Elevada Normal GALT Elevada GALE Normal - Elevada n Actividad Enzimática: GALT

Diagnóstico Neonatal Sistemático del Recién Nacido n UMTEST GAL de SUMA: usado en CSSL

Pesquisa Neonatal: medición de Galactosa TOTAL

Pesquisa Neonatal
 n Gal-1 -P= Gal Total (2º) – Gal libre (1º) n")
Gal-1 -fosfato (mg/dl) n Gal-1 -P= Gal Total (2º) – Gal libre (1º) n % Gal-1 -P= [Gal-1 -P]. 100 [Gal Total]

TP: KIT
 en eritrocitos")
Determinación Enzimática n Galactosa 1 - Fosfato Uridil Transferasa (GALT) en eritrocitos

Basados en la Reacción de Beutler Opción 1 Opción 2

Ausencia NADPH – No hay decoloración del Azul de Metileno NADPH actividad de GALT rapidez de decoloración de AM

SIGMA

n Análisis Molecular: n Detección de Mutaciones por amplificación por PCR+ER

Mutación Q 188 R Mutación N 314 D Mutación S 135 L En Brasil la galactosemia tiene una frecuencia de 1: 19. 984 recién nacidos. Q 188 R: exon 6, Por PCR se amplifica PA: 256 bp. El cambio de de una A por G crea un sitio de corte para la enzima de restricción Hpa. I. El alelo Q 188 R genera dos bandas de 158 y 98 bp. El alelo normal tiene un fragmento de 256 pb. n N 314 D, Duarte variant: exon 10, Por PCR se amplifica PA: 166 pb La mutación genera un sitio de corte para la enzima Ava II El alelo N 314 D genera dos bandas de 100 y 66 bp. El alelo normal tiene un fragmento de 166 pb. n S 135 L: exon 5, Por PCR se amplifica PA: 252 bp. La mutación genera un sitio de corte para la enzima Taq. I en el exón 5. El alelo S 135 L genera dos bandas de 192 y 60 bp. El alelo normal tiene un fragmento de 252 pb. n
 Q 188 R – exon 6 GALT gene. L, marker; lane 1, mutated")
(A) Q 188 R – exon 6 GALT gene. L, marker; lane 1, mutated homozygous (referred case); lanes 2 and 3, heterozygous (two screened cases) and lanes 4– 10, homozygous wild-type allele. Q 188 R (+) Hpa (B) N 314 D – exon 10 GALT gene. L, marker; lane 4, homozygous mutated allele (referred case); lanes 1, 2, 5 and 6, heterozygous and lines 3, 7– 10, homozygous wild-type allele N 314 D (+) Ava. II After amplification, the PCR products were digested overnight at 37 -C using the enzymes Hpa. II for exon 6, Ava. II for exon 10 and Taq. I for exon 5.

Patogenia I: deficiencia de GALK n n La deficiencia de GALK impide la fosforilación de galactosa a Gal-1 -P, lo que supone un acúmulo de galactosa en sangre, y la consiguiente producción de galactonato y galactitol El acúmulo de galactitol produce edema de las fibras del cristalino, y una desnaturalización de las proteínas, responsable del desarrollo de la catarata.

Aumento de Galactitol n Catarata nuclear “en gota de aceite” que puede tener un inicio intrauterino, en casos excepcionales

Patogenia II: déficit GALT El déficit GALT se produce un aumento de Gal-1 -P eritrocitaria y de la galactosa plasmática con la consiguiente galactosuria n La galactosa en exceso en el plasma es convertida en galactonato, y en galactitol. n

Patogenia II: Metabolitos tóxicos Gal-1 -P: - Pi y ATP AMP deaminasa Ac. Urico; n Inhibición de enzimas. - Inhibe glu-6 -fosfatasa y glucógeno fosforilasa hipoglucemia - Inhibe la función inositol monofosfatasa inositol en cerebro. n Balance UDP-Gal / UDP-Glu: actividad galactosil-transferasa n

Pi ADP AMP
 ADP y AMP Pi (80%) (-) inositol monofosfatasa inositol en cerebro.")
ATP (40%) ADP y AMP Pi (80%) (-) inositol monofosfatasa inositol en cerebro. (-) actividad galactosil-transferasa

Patogenia III: déficit GALE En el déficit GALE se produce un acúmulo de UDPGal e incluso de Gal 1 -P n Hay un déficit en la síntesis endógena de galactosa y en la producción de galactolípidos y galactoproteínas n

Determinación de metabolitos y actividad enzimática: n Gal-1 -P en eritrocitos n Gal TOTAL en eritrocitos Tabla VIII: Resumen de los niveles de metabolitos Deficiencia Galactosa (Plasma u Orina) Gal-1 -P (sangre) GALK Elevada Normal GALT Elevada GALE Normal - Elevada n Actividad Enzimática: GALT

Pesquisa Neonatal n Para la determinación de Fenilcetonuria y Galactosemia, el niño debe estar recibiendo alimentación láctea (materna o artificial), durante por lo menos 24 horas al momento del examen. Si el recién nacido está siendo alimentado con fórmulas especiales (enteral y/o parenteral), es necesario dejar constancia de ello por escrito, en la tarjeta recolectora
, DG? ?")
Formas de informar resultados: ? ? Duarte 2/Duarte 2 (homocigota), DG? ?

Diciembre 2010 con un total de 643. 409 RN n n 15 pacientes con Galactosemias Clásicas (Frecuencia 1: 42. 894), 44 pacientes con Deficiencias Parciales de GAL-T (Frecuencia 1: 14. 623), 3 pacientes con Deficiencias de GAL-K (Frecuencia 1: 214. 470) y 2 pacientes con Deficiencias de GAL-E (Frecuencia 1: 321. 705). Otros países: 1: 68. 400
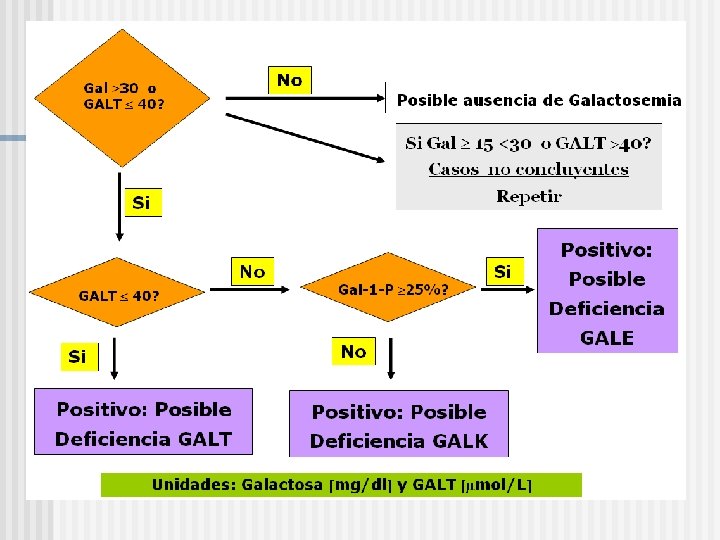
 GN")
Algoritmo (Manual de Procedimientos del Ministerio de Salud de la Nación, página 57) GN DN DD (depende: D 1 D 1, D 2 D 2 ó D 1 D 2? ?

Tratamiento n n n La galactosa de la dieta debe ser eliminada ante la menor sospecha La ingesta diaria no debería contener nunca más de 125 mg de galactosa (frente a los 6. 500 mg que por término medio tiene una dieta de un adulto normal); Una dieta estricta contiene aproximadamente unos 40 mg

Alimentos lácteos n n La leche y todos sus derivados son la principal fuente de galactosa liberada en el intestino mediante hidrólisis de la lactosa; y por tanto deben ser excluidos de la dieta en cualquiera de sus formas. La caseína es probablemente también una fuente importante de galactosa libre, ya que 100 gr. aportan 184 mg, y una fórmula láctea cuyas proteínas sean un hidrolizado de caseína, puede llegar a tener 60 -75 mg por litro.
")
Tabla IV: Contenidos aproximados en galactosa soluble de algunos alimentos (mg/100 gr de alimentos) Menos de 5 mg Melón, uva, pomelo, naranja, fresas Espárragos, remolacha, repollo, coliflor, apio, pepino, berenjena, espinaca, lechuga y maíz. Entre 5 -10 mg Manzana, banana, pera. Brócoli, zanahoria, cebolla, nabo. Entre 10 -20 mg Kivi, sandia, piña. Col de Bruselas, calabazas, sandia, batata. Entre 20 -30 mg Arándano Tomate Entre 30 -50 mg Soja, alubias Más de 100 mg Lentejas, guisantes, porotos Más de 400 mg Higos secos, pasas, avellanas Garbanzos

Control del tratamiento: n n n Los marcadores utilizados para el seguimiento del tratamiento dietético son los niveles de Galactosa-1 -P eritrocitarios, y galactitol plasmático. Valores de 4 mg % de Gal-1 -P, y de Valor máximo aceptable 25 -30 mol/L de galactitol plasmático (mg/dl =38 x mol/L).

Resultado Pesquisa Neonatal gal y enzima GALT o gal y GALT: N GALT< 1% GALT 1 -10% GALT 10 -75% Gal-1 -P N/ Gal-1 -P Mutación GALT presente GALT: N Gal-1 -P: N Galactitol GALT: N Gal-1 -P Galactitol Normal Actividad GALK Variante/ Galactosemia Variante de Deficiencia Benigna o Clásica GALK Galactosemia Portador Galactitol Actividad GALE Falso Positivo Deficiencia GALE

n Alguna pregunta?
- Slides: 63