Qumica Biolgica Patolgica ALTERACIONES EN EL METABOLISMO DE

Química Biológica Patológica ALTERACIONES EN EL METABOLISMO DE LAS LIPOPROTEINAS- I - 2016 Tema: 13 Dra. Silvia Varas svaras @unsl. edu. ar

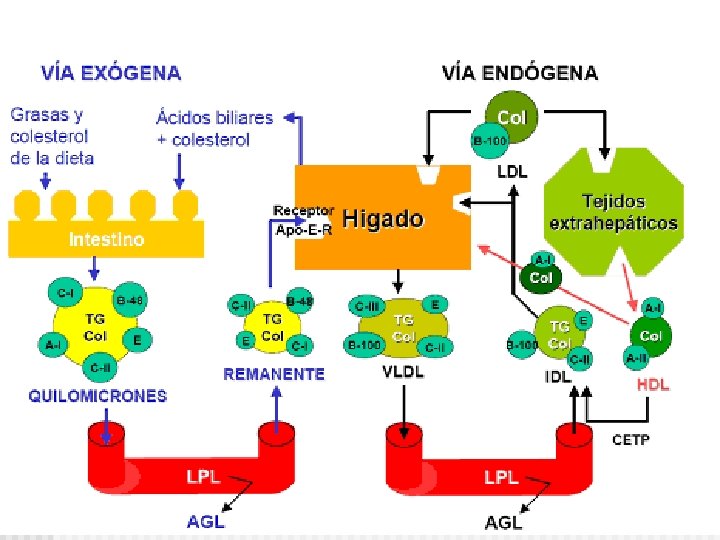

Resumen Metabolismo de Lipoproteínas:
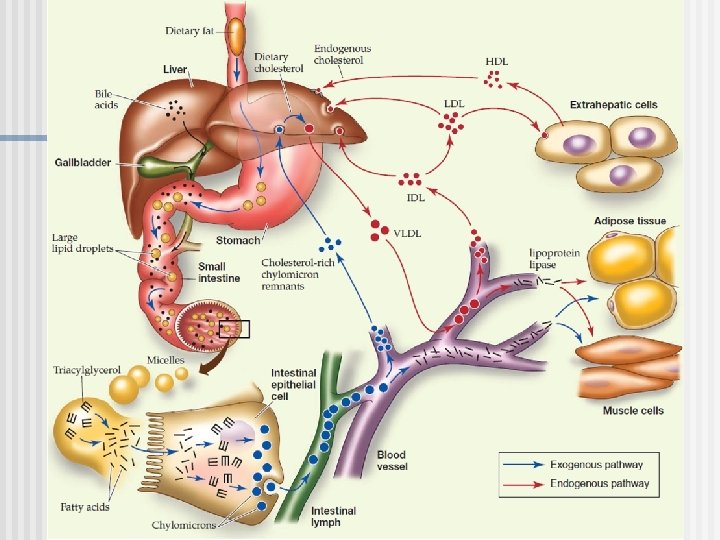

Dr. Fernando D. Brites, 2016. ABSL

Menores de 20 años Dr. Fernando D. Brites, 2016. ABSL

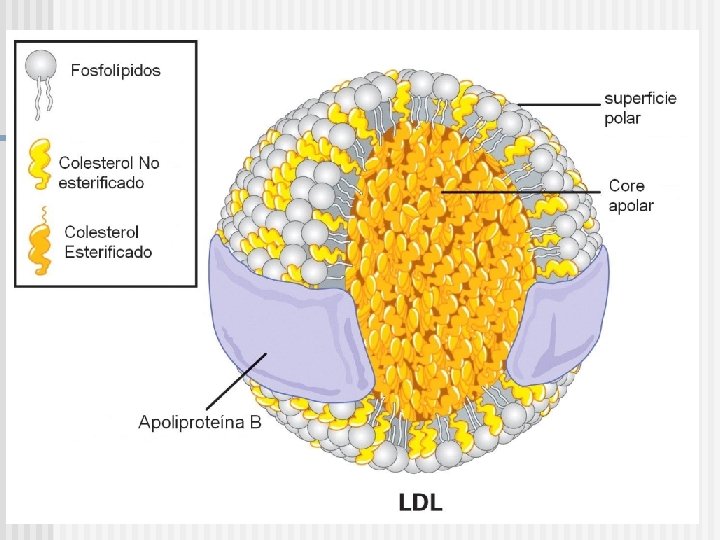
Estructura de Lipoproteínas plasmáticas n LP core: Triglicéridos n Esteres de colesterol n n LP superficie: Fosfolípidos n Proteínas n Colesterol Libre n

Triglicéridos Protagonistas I: Lípidos Fosfolípidos Colesterol Ésteres de Colesterol FL: Lecitina

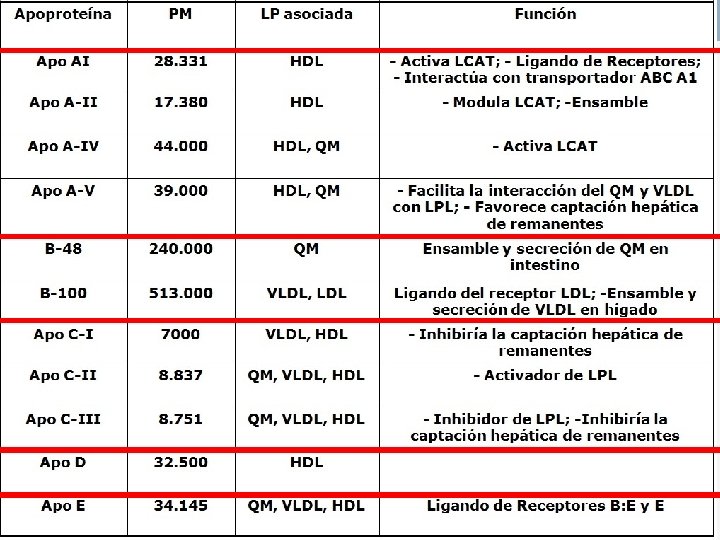
Protagonistas II: Apoliproteínas Las Apolipoproteínas, varían de unas lipoproteínas a otras. Se denominan con letras del alfabeto, de acuerdo con el orden en el que han sido descubiertas. n Las principales son Apo A, Apo B, Apo C, Apo D, Apo E. Son muy importantes y con distintas funciones: - Mantienen la integridad estructural de las lipoproteínas - Regulan las enzimas que intervienen en el metabolismo de las LP. - Actúan como ligandos para reconocer receptores específicos. n
,")
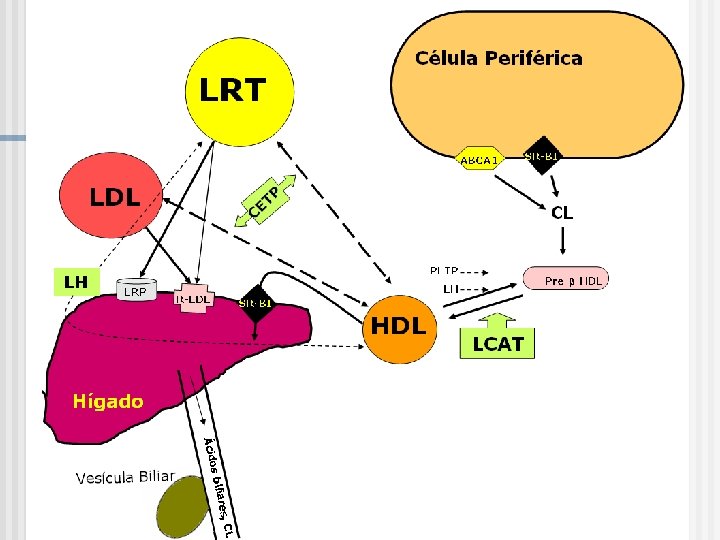
Protagonistas III: RECEPTORES DE LIPOPROTEÍNAS n Los receptores HEPATICO: R-LDL (Apo E: B 100), los LRPs (proteína relacionada con el receptor de LDL); y receptor de HDL 2 (SR-B 1, scanvenger receptor B 1). n A nivel EXTRAHEPATICO, existen receptores de LDL (B 100), LRPs y los basureros SR-A (para LDL alteradas: acetiladas, oxidadas o glicosiladas) presentes en los macrófagos y los SR-B 1.
,")
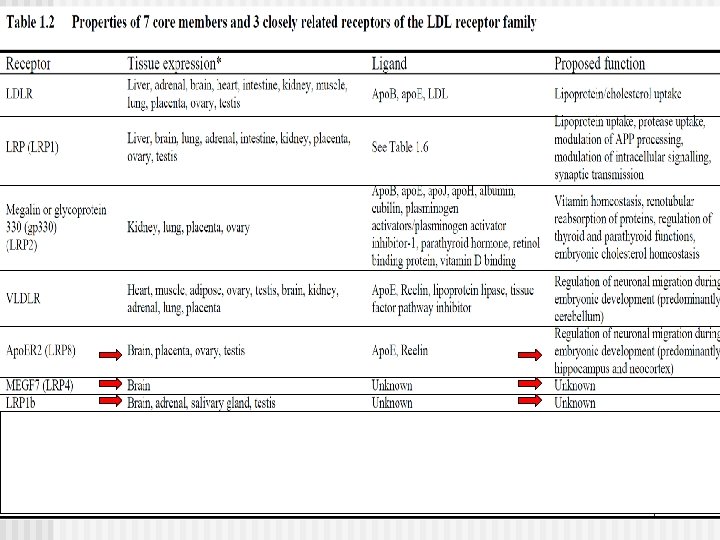
Familia Receptor LDL La flia comprende 7 miembros: n RLDL n LRP (LRP 1), n LRP 1 b, n LRP 2 (también llamada MEGALINA), n LRP 4 (también llamada MEGF 7 (multiple epidermal growth factor-like domains protein 7), n LRP 8 (también llamada Receptor apolipoprotein E 2), n Receptor VLDL receptor (VLDLR)

n n n MEGF 7 LRP 8 LRP 2

Protagonistas IV: Sistemas Enzimáticos Lipoprotein lipasa periférica, es sintetizada en las células, translocada a la superficie de la pared vascular y liberada por la heparina. Es activada por la Apo C 2 e inhibida por la Apo C 3 y es sensible a la insulina. Es responsable de la catabolización de quilomicrones y VLDL. Lipasa hepática, está regulada por la síntesis de colesterol a nivel hepático, es responsable del catabolismo de los QMr, IDL y HDL 2. Lecitin colesterol acil transferasa (LCAT), esterifica el colesterol libre en las HDL. Transfiere ácidos grasos desde los fosfolípidos al colesterol libre. Es estimulada por la Apo A 1 y Apo. E. Proteína transportadora de colesterol esterificado (CETP) es responsable del transporte de los ésteres de colesterol desde las HDL VLDL, IDL y LDL y de triglicéridos desde las VLDL HDL.
: Enzima S Función Origen (+) (-) LPL")
Protagonistas IV: Sistemas Enzimáticos (+ y -): Enzima S Función Origen (+) (-) LPL TG y FL de los QM, VLDL TG hidrolasa FLipasa Tej adiposo y musculo esqueletico y cardiaco Apo CII *Insulina Heparina LH TG y FL de los IDL, HDL >TGhidrolasa < FLipasa Hepatocitos Insulina Estrógenos Andrógenos SDS HORMONAS TIROIDEAS Heparina Apo CIII Protamina -Bloquean.
: Enzima S Función Origen CETP * CE")
Protagonistas IV: Sistemas Enzimáticos (+ y -): Enzima S Función Origen CETP * CE Transfiere CE por TG de HDL a LP con Apo B Hígado y Tejido Adiposo (TA) PLTP * FLs y CL de QMr y VLDLr Transfiere FL hacia la HDL desde los remanentes (Remodelamiento de HDL) Hígado, TA y Pulmón LCAT * CL y Fosfatidil- colina de las HDL Esterificación Hepatocito del colesterol * Circulan unido a HDL (+) Apo AI Apo E (-) -

Funciones PLTP

PLTP n n Circula unido a HDL y media la transferencia neta de fosfolípidos hacia la HDL y también intercambia fosfolípidos entre lipoproteínas. La transferencia neta de fosfolípidos en la HDL resulta en la formación especies menos densas y mas grandes. Es capaz de transferir todos los FL incluidos fosfolípidos, diacilglycerol, α-tocoferol, cerebrósidos, etc. Transferencia neta de colesterol libre hacia la HDL

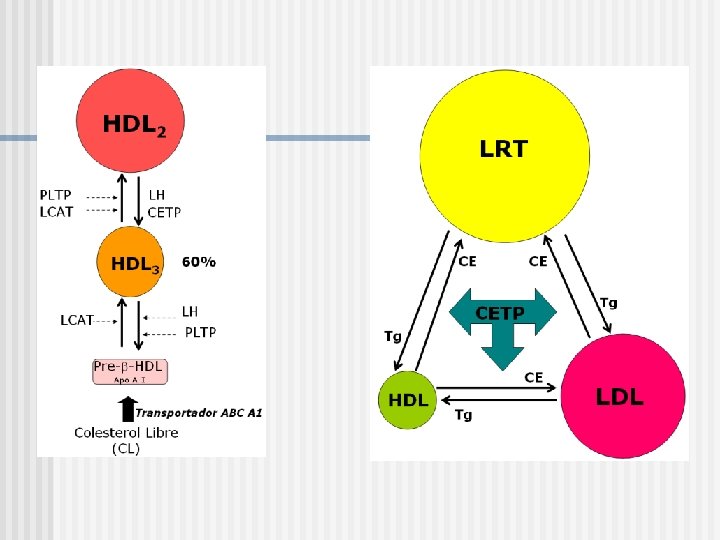
HDL 2

El reemplazo de apo. A-I con apo. A-II genera un efecto inhibitorio sobre PLTP. La conversión de HDL generates HDL 2 y prebeta -HDL. Un Tg en las HDL aumenta el proceso de conversión.
 Apo A-I PLTP CL LH LCAT HDL 3 CETP LH LCAT")
Colesterol Libre (CL) Apo A-I PLTP CL LH LCAT HDL 3 CETP LH LCAT PLTP HDL 2 Fragmentos de Superficie (CL, FL) Pre- -HDL LPL LRT

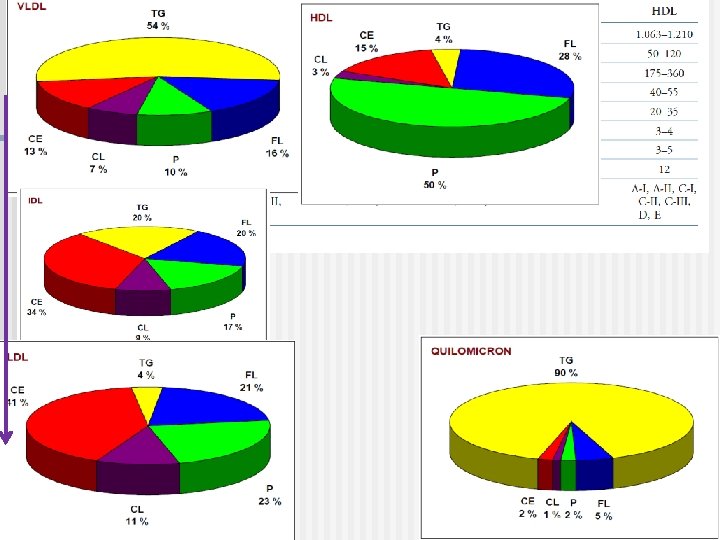
P 100% C P P Composición 80% C P 60% C T 40% C T 20% T T LDL HDL 0% Q VLDL Tipo de LP Figure 2. LP y su contenido relativo de triaglicéridos(T), colesterol (C) y proteinas (P).

Microfotografía de las lipoproteínas
 Origen y función")
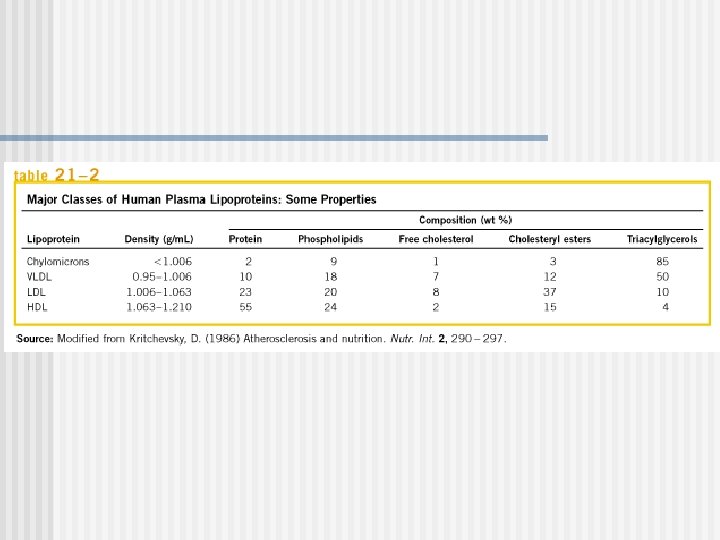
Las cinco clases de Lipoproteinas Aumenta la densidad Clase Diámetro (nm) Origen y función Principal apoliproteínas Quilomicrones (QM) 500 Intestino. Transporte de TAG dietarios A, B 48, C(I, III) E Very low density lipoproteins (VLDL) 43 Hígado. Transporte de Tg endogenamente sintetizada TAG B 100, C(I, III) , E Low density lipoproteins (LDL) 22 Formada es circulación por ruptura parcial de IDL. Libera colesterol a los órganos perisfericos B 100 High density lipoproteins (HDL) 8 Hígado. Remueve colesterol “usados” de los tejidos y es llevado al hígado. Dona apolipoproteinas a QM y VLDL A, C(I, III), D, E

- densas + densas
 (+)")
LIPIDOGRAMAS β Pre-β α ELECTROFORÉTICOS siembra (-) (+)

 apolipoproteinas: A 1,")
OBJETIVOS: 2. Definirlas funciones en el transporte y procesamiento de: a) apolipoproteinas: A 1, B 48, B 100, CII, y. E; b) enzimas: lecitin: colesterol acil transferasa (LCAT), acil. Co. A: colesterol acil transferasa (ACAT) y lipoprotein lipasa (LPL); c) proteínas unicas: triagliycerol transfer protein (MTTP), ABC A 1 y NPC-1 Enzima (Proteína) Sitio de Acción Activador Función b) LPL Vasos sanguíneos Apo CII b) ACAT Interior de las células Colesterol Libre Escinde AGL desde en QM y VLDL para ser usado por TA y músculo Acumulación de esteres de colesterol b) LCAT Sangre Apo A 1 c) ABC-A 1 Membrana plasma Apo A 1 c) MTTP 1 REL intestino/hígado Ninguno a) Apo A 1 Ninguno a) Apo B-48 Sangre, membrana plasmática intestino Ninguno Activa LCAT y ABC 1; une a receptores SR-B 1 sobre las células que requieren extracción de colesterol Exporta de QM desde células intestinales a) Apo B 100 Hígado, otras células Ninguno Ligando para R de LDL; exporta VLDL desde el hígado a) Apo CII Vasos sanguíneos Ninguno Activa LPL a) Apo E Hígado Ninguno c) NPC-1 2 Interior de las células Ninguno Ligando para el receptor de apo E que depura los QMr, IDL y HDL Transfiere el Colesterol tomado de las LDL de endosoma-lisosoma al pool celular en el Golgi 1 -Microsomal Triacylglycerol Transfer Protein Extracción de Colesterol desde células HDL transporta CE para depuración por el hígado por medio de la conversión a A. Biliares Transporta colesterol y FL( lecitina) hacia fuera de la bicapa lipídica hacia apo proteinas como Apo A 1 para generar la pre beta HDL Carga TG en B 48 (intestino) y B 100 (hígado) 2 - Niemann Pick C Protein -1
 Def. LPL: 1 en 1. 000 Def. Apo C 2:")
Clasificación de Fredrickson (OMS) Def. LPL: 1 en 1. 000 Def. Apo C 2: 1 en 1. 000 Aa: 1 en 500 AA: 1 en 1. 000 1 en 100 1 en 5. 000 HTF: 1 en 100

Deficiencia Fliar LCAT
 Déficit Apo CII")
Clasificación General VIA EXOGENA þ þ Déficit en LPL (Tipo I) Déficit Apo CII (Tipo Ib) þ Hiper þ þ VIA ENDOGENA þ þ Hipo þ þ VIA EXOGENA y ENDOGENA þ þ Hipertrigliceridemias endógena Hiperlipidemia familiar combinada Hipercolesterolemia familiar Apo B 100 defectuosa familiar Hipercolesterolemia Poligénica Abeta Lipoproteinemia Hipobetalipoproteinemia familiar Déficit Lipasa Hepática Disbeta. Lipoproteinemia (Tipo III, Beta ancha) þ Deficiencia HDL þ Alteraciones Familiares del Metabolismo HDL þ VIA REVERSA þ þ þ Hipo- -lipoproteinemia familiar Deficiencia LCAT (ojo de pez) Enfermedad de Tangier Deficiencia Apo AI Hiper- -lipoproteinemia Deficiencia CETP

Tabla Nº 4 : Síntomas y hallazgos de laboratorio Entidad Sintomatología Tg CT HDLc Déficit de LPL Dolor abdominal, pancreatitis, xantomas, lipemia retinalis Déficit de Apo C-II Dolor abdominal, pancreatitis, xantomas, lipemia retinalis Hipertrigliceridemia familiar Dolor abdominal, pancreatitis y xantomas N, FCH Hipertensión, obesidad, resistencia a la insulina. Estenosis vascular, infarto<50 años. FH(homocigota) Xantomas, arco corneal, estenosis vascular, infarto muy precoz. N FH (heterocigota) Xantomas, estenosis vascular , infarto<50 años N Apo B-100 defectuosa Xantomas, estenosis vascular, infarto<50 años N N, Abetaliproteinemia Malabsorción, ataxia, retinitis pigmentaria Hipobetaliproteinemia (homocigota) Malabsorción, ataxia, retinitis pigmentaria Hipobetaliproteinemia (heterocigota) Acantocitosis, esteatosis hepática, retinopatía, diabetes alteraciones neurologicas N N Déficit de LH Estenosis vascular, infarto<50 años Disbetalipoproteinemia Xantomas, estenosis vascular, infarto<50 años , N N Déficit de LCAT (parcial) Arco corneal, depósitos corneales <20 años N, N Déficit de LCAT(total) Arco corneal, depósitos corneales <20 años, insuficiencia renal Enfermedad de Tangier Amígdalas naranjas, esplenomegalia, ateroclerosis precoz , N Déficit de Apo A 1 Estenosis vascular, infarto<50 años , N N Referencias: N: normal; : elevado; : muy elevado; : bajo; : muy bajo

Síndrome de Quilomicronemia n Deficiencia familiar de LPL n Deficiencia familiar de Apo C-II

Estructura y Función de LPL Modelo esquemático de acción de LPL en el endotelio capilar: Los proteoglicanos como el heparan sulfato son intercalados sobre el endotelio capilar. LPL se unen al heparan y se extienden hacia el lumen vascular Heparan sulfato

N-glicosilación: Asn 43 y Asn 359. La sustitución de N 43 por A suprime completamente la actividad y resulta en la acumulación de la proteína inactiva en el ER mutación Lisosomas Procesamiento postranscripcional y extracelular de LPL: LPL es sintetizada en las células del parénquima en forma inactiva. La activación de la enzima ocurre en el aparato de Golgi. La enzima activa es liberada de la célula y se une a la superficie de la célula endotelial. La enzima inactiva se puede liberar desde la célula

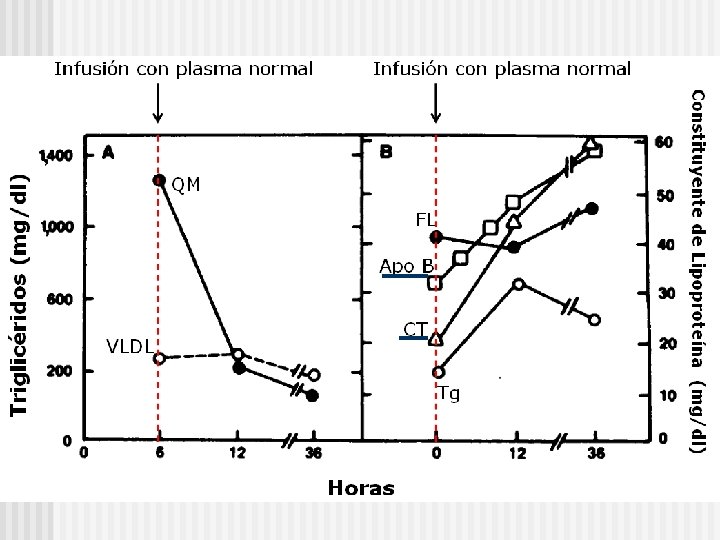
Metabolismo de los QM

Quilomicrones n n Sintetizados en ID Transporte de lípidos dietarios 98% lipidos, mayor tamaño, baja densidad Apo B-48 • Receptor binding n Apo C-II • Activador de Lipoprotein lipasa n Apo E • Remanente Receptor Binding

- La LPL madura funcional es un homodímero. - Activación por glicosilación en Asparagina y dimerización en el RE. - LPL es primero liberada a las vesículas secretoras (VS) y luego a los lisosomas para la degradación intracelular o a la superficie celular del parénquima, donde la enzima se une a PG-HS. - Finalmente, LPL se traslada a los sitios de unión de Heparan Sulfato funcionales en la superficie luminal del endotelio capilar, donde lleva a cabo la hidrólisis de las lipoproteínas ricas en TGs. Glycosyl phosphatidyl inositol-anchored high density lipoprotein- binding protein 1 (GPIHBP 1) es una nueva molécula en la célula endotelial y juega un rol en importante en metabolismo de los QM. Animales deficientes en GPIHBP 1 presentan hiperquilomicronemia

GPIHBP 1: glycosylphosphatidylinositol-anchored HDL-binding protein 1 El primer paso en el catabolismo de lipoproteínas ricas en triglicéridos (LRT) ocurren en tejido adiposo blanco y músculo estriado. Los dímeros de GPIHBP 1 proveen el sitio de anclaje sobre la superficie del endotelio capilar. El endotelio luego libera las lipoproteínas remanente que vuelve a la circulación

Deficiencia Familiar de LPL Descripta por 1ª vez en 1932 por Bürger and Grütz, en un niño de un matrimonio consaguineo. n En 1960, Havel y Gordon demuestran un clearence defectuoso de las LP ricas en triglicéridos. n En 1974, Kraus y col. informaron la actividad disminuida de LPL y LH, en plasma postheparinico. n En 1989, se describe el primer defecto en la estructura del gen de LPL. n Frecuencia: 1/1. 000 n

 QM son extremadamente elevados luego del")
Características: Lípidos de la sangre y lipoproteínas: a) QM son extremadamente elevados luego del nacimiento. De 43 casos, 13 fueron detectados durante el primer año de vida, 22 (antes de 10 años). b) VLDL, HDL son normal, pueden ser bajos. c) [TG] estan elevados: 2. 500 -12. 000 mg/dl. Normal: 100 -250 mg/d. L. d) [Colesterol] es alta, pero no extremadamente alta. e) Capa cremosa en plasma enfriado a (4 o. C, toda la noche).
 de la piel y")
Fenotipo Clínico n n n Los xantomas (tipo papulo-eruptivos) de la piel y la lipemia retinalis aparecen con niveles de tg≥ 2. 000 mg/dl y pueden desaparecer en varias semanas si los niveles de tg disminuyen. Los xantomas eruptivos son nódulos pequeños y amarillos que se elevan en la piel (con una área pigmentada de color rojo que lo rodea). Los xantomas eruptivos se observan en nalgas y zonas extensoras

xantomas eruptivos El examen del fondo de ojo, donde aparece una imagen típica, la Lipemia Retinalis, caracterizada por vasos de coloración rosa pálido debido al aumento de los quilomicrones.

n Se observan los vasos retinianos de color blanquecino y un fondo de ojo de aspecto rosa pálido

Fenotipo Clínico II n n n Organomegalia: particularmente, hepatoesplenomegalia Presencia de células espumosas. Dolor en abdomen agudo Riesgo de pancreatitis recurrente con evolución a crónica. Genética: El gen de la LPL, esta situado en el cromosoma 8 p 22.

Pancreatitis La teoría más extendida sostiene que la acción de la lipasa pancreática en un plasma con exceso de TG provoca la acumulación de ácidos grasos en el tejido pancreático. n Estos, mediante la producción de radicales libres, dañan el páncreas tanto a nivel acinar como vascular muerte celular por apoptosis. n

Presentan tres motivos que interactúan con los factores de transcripción Oct 1, C/EBP (CAAT enhancer-binding protein) y Sp 1, ubicado dentro de 100 pb upstream del sitio de inicio de la transcripción, donde se ha demostrado que es un sitio crítico para la actividad basal del promotor Gen LPL Fibratos y las Tiazolidinas (+)

Gen LPL y Mutaciones 1º Doble heterocigota 8 p 22

40 mutaciones 28 mutaciones con cambio de sentido asociadas con reducida o ausente actividad de LPL y una variante con actividad LPL normal, 7 sustituciones puntuales que causan un codon stops (sin sentido); 2 mutaciones en el marco de lectura; y aparte de las dos mutaciones descriptas originalmente, Pocas deleciones de 2, 3 y 6 Kb 2 mutaciones en el sitio del splicing
 Localización de mutaciones con cambio de sentido en el gen")
G 188 E (40%) Localización de mutaciones con cambio de sentido en el gen LPL. Solo 5 de los 10 exones se muestran con detalle

DIAGNÓSTICO BIOQUIMICO Actividad de lipasa moles de glicerol/m. L/hr : LPL Lipasa Extrahepatica Hepatica Normal Def. Hombre: 4 -7 0. 5 15. 4 13. 5 Mujer: 4 -7. 5 0. 1 11. 6 11. 2
![Diagnóstico [TG] de plasma [quilomicrones] Responde a la restricción en grasa < 20 g/día](http://slidetodoc.com/presentation_image/327ea1938ef6c47d69ce2c136da62354/image-62.jpg "Diagnóstico [TG] de plasma [quilomicrones] Responde a la restricción en grasa < 20 g/día")
Diagnóstico [TG] de plasma [quilomicrones] Responde a la restricción en grasa < 20 g/día TG < 400 mg/d. L Col < 250 mg/d. L Medida actividad de LPL Capa blanca en plasma dejado a 4 o. C toda la noche.

Tratamiento: Resto de su vida: dieta restringuida en grasa Pequeñas cantidades de AG esenciales. Se les suministró TG de cadena media(TGCM) puede ser bien tolerada. TGCM contiene solo 812 carbonos. Son transportado al hígado vía vena porta. Esto AGCM son captados por las células por transportador especifico CD 36(FAT), no son reesterificados a triglicéridos, y, lo más importante, no son empaquetados en QM.
iso-BCFA Palmitoleico Oleico 16 D 9(ω7)")
AG Insaturados AG Saturados Acetico 2 Monoinsaturados Propinoico 3(OCFA)iso-BCFA Palmitoleico Oleico 16 D 9(ω7) 18 D 9(ω9) Butirico 4 Valerico 5(OCFA)iso-BCFA Erucico Caproico 6 Nervonico Caprilico 8 Poliinsaturados Caprico 10 Linoleico 18 D 9, 12(ω6) Laurico 12 Miristico 22 D 13(ω9) 24 D 15(ω9) 14 Palmitico 16(25%) α- linolenico 18 D 9, 12, 15(ω3) (γ-D 9, 12, 6(ω6)) Estearico 18(5%) Araquidonico 20 D 5, 10, 11, 14(ω6) Araquidónico 20 Timnodonico 20 D 5, 8, 11, 14, 17(ω3)EPA Lignocérico 24 Clupanodonico 22 D 7, 10, 13, 16, 19(ω3)DPA Cervonico 22 D 4, 7, 10, 13, 16, 19(ω3)DHA

Deficiencia Familiar de Apo CII

Historia n n 1978: Breckenridge y col. Informan el caso de un hombre de 59 años de edad que desde los 18 años había tenido dolor abdominal y había desarrollado diabetes mellitus y esteatorrea. Con una dieta baja en grasa sus triglicéridos descendían de 4000 a 1000 mg/dl. Al darle una transfusión por una anemia, observó que el defecto corregía, sugiriendo que un factor presente en el plasma estaba deficiente. Luego se comprobó que tenía un déficit de Apo CII.

GPIHBP 1: glycosylphosphatidylinositol-anchored HDL-binding protein 1

Diferencia con Deficiencia de LPL: Se diferencia de deficiencia familiar de LPL en que: n (1) los síntomas generalmente se desarrollan a una edad más avanzada (13 -60 años) y n (2) los individuos pueden desarrollar insuficiencia pancreática crónica con esteatorrea y diabetes mellitus dependiente de insulina.

Estructura del Gen n Encontraron que el gen APOCII bandea en el cromosoma 19, la banda 19 q 13. 3. Esta constituido por cuatro exones.

Tipos de Mutación n Se han identificaron múltiples mutaciones de cambio de una base por otra que genera un codon stop y deleciones e inserciones que generan mutaciones en el marco de lectura en el gen APO CII

Diagnóstico: n n El diagnóstico se basa en la determinación de la concentración de apo C-II. Disminución de la actividad de LPL en ausencia de Apo CII y corrige a valores normales con la adición de Apo CII normal.

Tratamiento: n El tratamiento es una dieta baja en grasas a lo largo de la vida. n Infusiones de plasma rico de Apo CII
- Slides: 73