PURPURA TROMBOCITOPENIC IMUN 1 Definiie PTI este cea

PURPURA TROMBOCITOPENICĂ IMUNĂ 1

Definiție • PTI este cea mai frecventă anomalie a hemostazei la copil • Hiperdistrucție realizată prin trombocitară mecanism periferică, imunologic, ce depășește capacitatea de trombocitopoeză compensatorie normală 2

Etiopatogenie • Mecanism imunologic: autoanticorpi ce reacționează cu glicoproteinele membranare de pe suprafața trombocitelor • Atc antitrombocitari din clasa Ig G se fixează pe suprafaţa trombocitelor şi se leagă prin porţiunea Fc de receptorul corespondent al macrofagelor sistemului reticuloendotelial splenic iar consecinţa este distrugerea trombocitului • Ac antitrombocitari sunt rareori din clasa Ig M 3

Clasificarea trombocitopeniilor imune PTI primare • forma clasică • forma legată de infecţia HIV PTI secundare • • LES boli limfoproliferative tumori solide indusă de medicamente indusă de infecţii purpura posttransfuzională (alloimună) trombocitopenia autoimună neonatală trombocitopenia alloimună neonatală 4

Aspecte clinice • debut la 2 -3 săptămâni după o intercurență infecțioasă o 80 -85% infecții virale: rujeolă, rubeolă, varicelă, parotidită epidemică, mononucleoză infecțioasă, gripă, parvoviroză, infecție HIV • manifestarea clinică caracteristică: purpura peteşială • echimoze • hemoragii la nivelul mucoaselor • foarte rar: hemoragii profunde intratisulare/cavitare 5
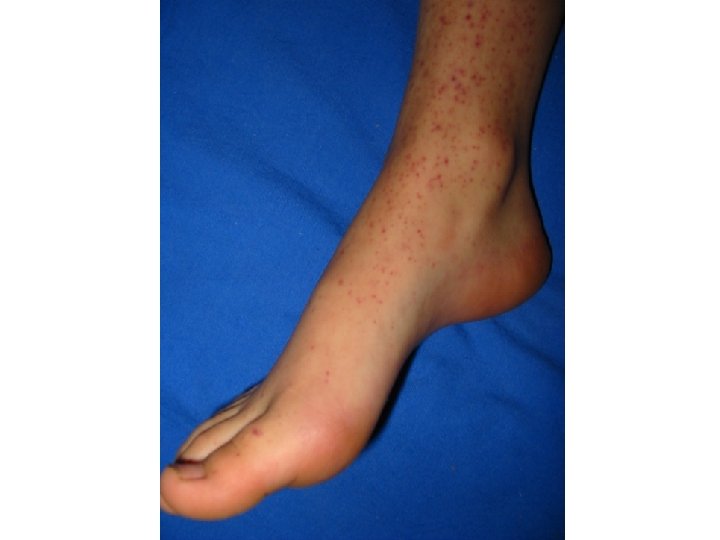
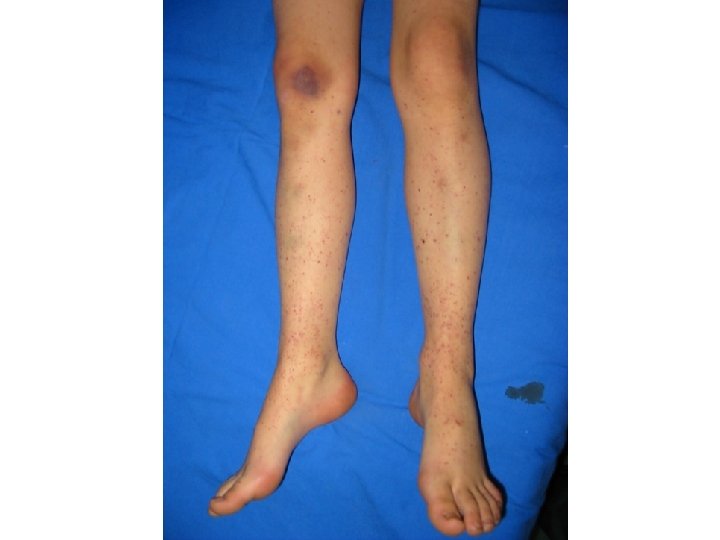
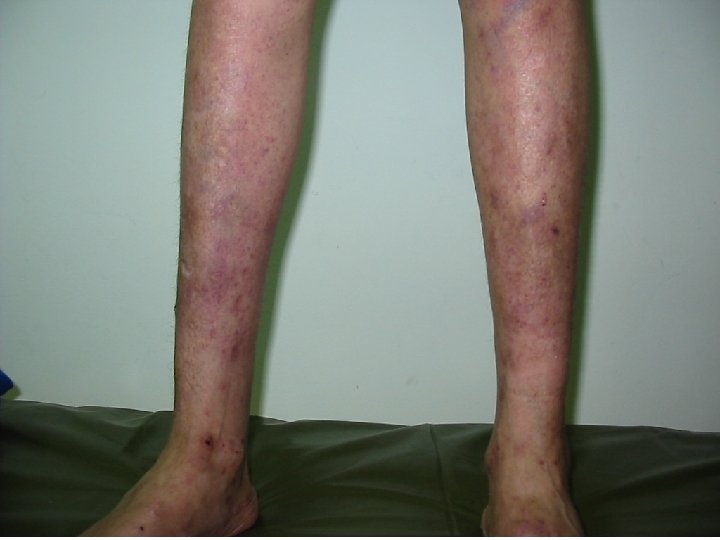
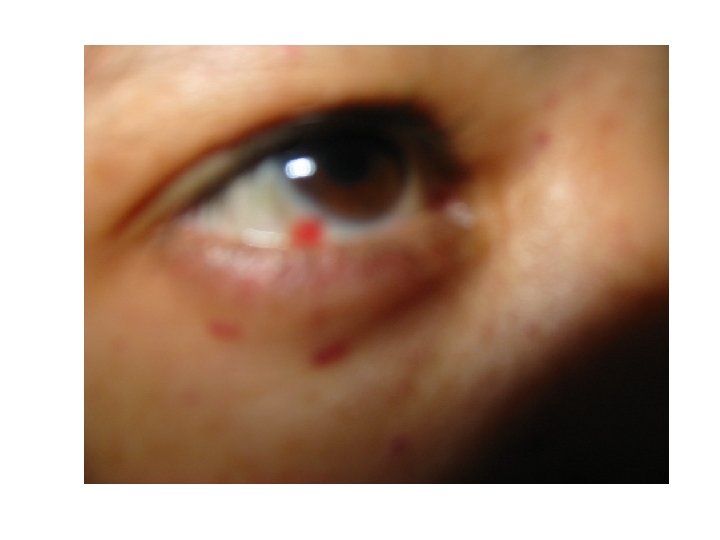
 Clasificare pe baza severității • Forme ușoare: Tr > 40.")
Aspecte clinice (cont. ) Clasificare pe baza severității • Forme ușoare: Tr > 40. 000/mm 3 (hemoragii prelungite după traumatisme) • Forme medii: Tr 20 – 40. 000/mm 3 (hemoragii spontane: peteşii cutaneo-mucoase, risc de hemoragii abundente: epistaxis, HDS) • Forme severe: Tr <20. 000/mm 3 (hemoragii spontane, risc de cerebrală) hemoragii fulminante şi de hemoragie 10
 –")
Aspecte evolutive PTI – Acute (75% din cazuri se vindecă în 6 luni) – Cronice – Recidivante Nu există criterii clinice sau paraclinice care să permită anticiparea evoluţiei acute autolimitate sau cronice 11

Diagnosticul PTI • Număr scăzut de trombocite • TS prelungit *Studiul Atc antitrombocitari este examinarea ideală dar este laborioasă şi costisitoare, motiv pentru care nu se practică de rutină. **Aspiratul osteo-medular are importanţă pentru excluderea debutului LAL, fiind esenţială efectuarea înainte de iniţierea corticoterapiei. 12

Tratamentul PTI Obiectiv: prevenirea şi combaterea hemoragiilor Nici una din metodele de tratament nu influenţează durata bolii dar există largi posibilităţi de creştere temporară a numărului de trombocite 13

Rolul tratamentului patogenetic Studiul Blanchette, Toronto, 1993 N 53 cazuri T 20. 000/mm 3 Tratament Durata Tr 50. 000/mm 3 Vindecare la 6 luni Ig. IV 2 zile 89% Prednison 4 zile 71% Fără 16 zile 81% 14

Tratamentul PTI acute Pentru reducerea duratei trombocitopeniei severe: • Ig. IV doză totală de 1 -2 g/kg • pulsterapie cu metilprednisolon, 1 g/m 2/zi, 1 -5 zile • prednison 2 mg/kg/zi, po, 10 -20 de zile 15
 Rare cazuri pot prezenta hemoragii severe ce nu pot")
Tratamentul PTI acute (cont. ) Rare cazuri pot prezenta hemoragii severe ce nu pot fi stăpânite cu mijloace obişnuite. Se recomandă atunci: • transfuzii de concentrat trombocitar : 0, 5 -1 U/m 2/oră • splenectomia “eroică” de urgenţă: numărul trombocitelor va creşte după 20 de minute de la pensarea pediculului splinei şi intervenţia poate fi salvatoare. • plasmafereza: rezultate inferioare celor raportate la adulţi în purpura trombocitopenică trombotică 16

Tratamentul PTI cronice 17
 • Splenectomia este metoda terapeutică de elecţie ce duce")
Tratamentul PTI cronice (cont. ) • Splenectomia este metoda terapeutică de elecţie ce duce la vindecare în 6588% din cazuri. 18

Splenectomia • După vârsta de 5 ani • Pregătirea preoperatorie: - Ig IV sau corticoizi vaccinare: antipneumococică, anti. Haemophilus, antimeningococică *Nu este necesară administrarea profilactică de transfuzii de trombocite. 19

Complicaţiile splenectomiei • infecţii cu pneumococul, bacterii incapsulate haemophilus ce includ influenzae şi meningococul Profilaxia acestor infecţii se poate realiza prin vaccinare, antibioprofilaxie sau tratamentul antibiotic al oricărui episod febril • trombocitoza cu risc de tromboze 20

Alternativele terapeutice în PTI cronică Sunt inferioare splenectomiei: • Corticoterapia în cură cronică • Alcaloizii de vinca: vincristina, vinblastina • Imunosupresoare: azathioprina, ciclofosfamida, ciclosporina, anticorpi anti CD 20 • Ig anti-Rh (D) 21

Mortalitatea în PTI • 0, 5 -1% prin hemoragie cerebrală • 1% prin septicemie postsplenectomie 22

TULBURĂRI ALE COAGULĂRII 23
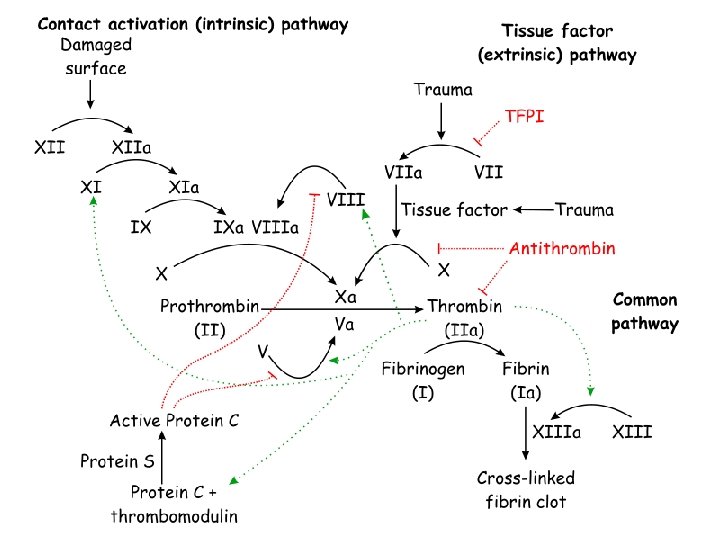

Schema coagulării Sistem intrinsec XII XI Sistem extrinsec VII IX VIII Xa V Protrombina II a Trombina Fibrinogen I FIBRINA 25

METODE DE EXPLORARE A HEMOSTAZEI Timpul vasculo-plachetar: TS, Tr Timpul plasmatic: - APTT, TC, TH: F XII, XI, IX, VIII, X, V, I, II -TQ/PT: F VII, X, V, II, I 26
 1. VASCULARE Purpura H-S N N N 2. TROMBOCITARE")
Tulburarea Tr TS TQ (PT) 1. VASCULARE Purpura H-S N N N 2. TROMBOCITARE PTI Trombastenia Efect aspirinic N N N 3. COAGULOPATII Hemofilia A Hemofilia B B. von Willebrand Deficitul de vit. K CID N N N N N N APTT N Alte N N N FVIII F IX Fv. W F II, VII, IX, X 27

Principalele coagulopatii din practica pediatrică Înnăscute • Hemofilia A • Hemofilia B • Boala von Willebrand Dobândite • CID • Deficienţa de vitamina K • Complicaţii hemoragice în boli hepatice • Inhibitorii coagulării 28

HEMOFILIA A • Hemofilia A este o coagulopatie ereditară X-linkată (gena situată pe brațul lung al cromosomului X) datorată deficitului de F VIII • Terapia actuală a transformat această boală potenţial letală sau grav invalidantă într-o afecţiune compatibilă cu o viaţă normală. • Incidenta: - 1: 5000 de băieti rasială sau etnică - 85% din cazurile de hemofilie sunt A 29

Efectele progreselor în genetică • Identificarea purtatoarelor genei • Diagnostic prenatal (amniocenteza, biopsia de vilozităţi coriale) • Producţia „industrială” de RHu FVIII • Terapia genică 30

Clasificare 31

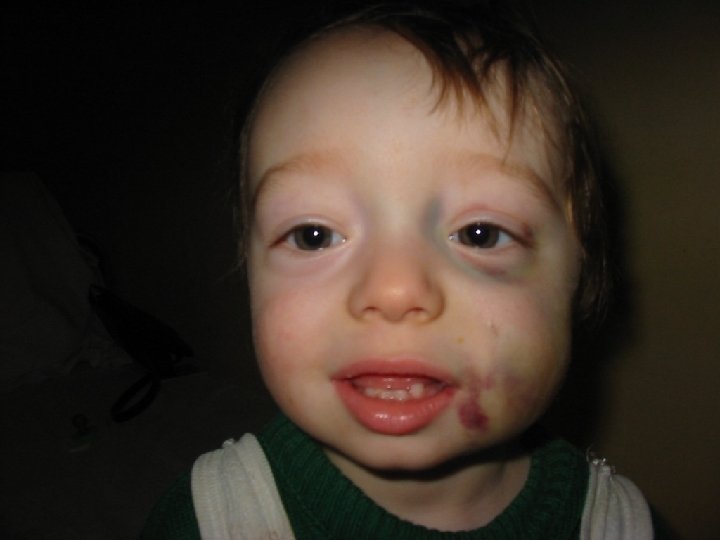
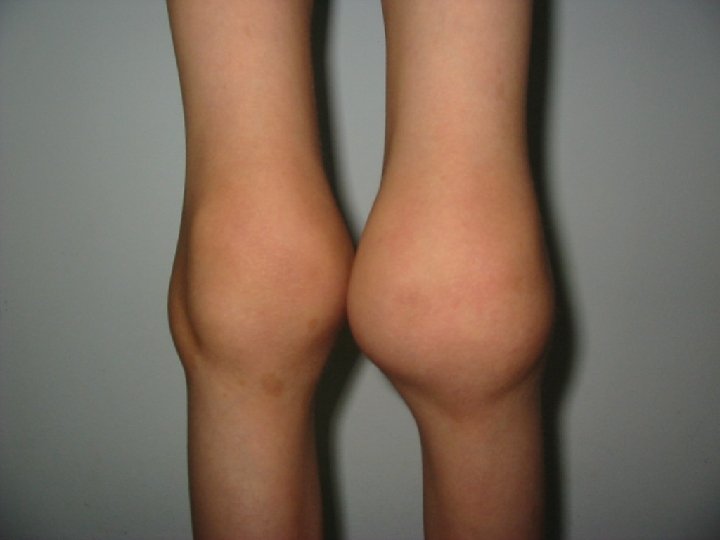
Debutul : Aspecte clinice • la naştere prin sângerare ombilicală, cefalhematom • cu ocazia circumciziei • cu ocazia erupţiei dentare • traumatisme legate de apariţia mersului Ulterior: • sângerări prelungite posttraumatice • hematoame profunde cu localizări bizare • hemartroze 32
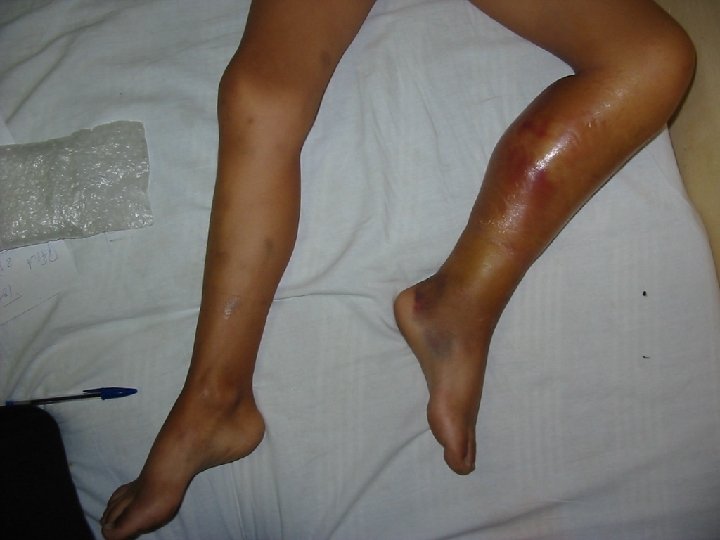
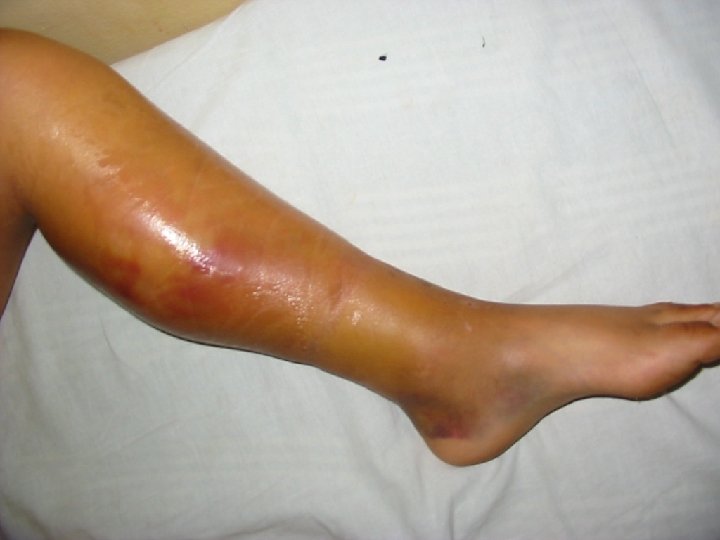

Hemoragie de psoas cu calcificări şi pseudotumoră care erodează aripa iliacă

Hemoragie intracraniană

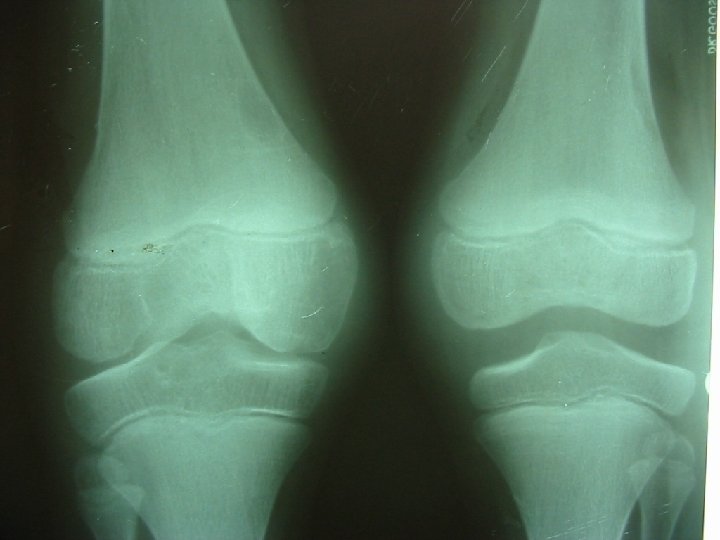
Atropatie hemofilică cu îngustarea spaţiului articular şi pseudochiste
 este prelungit *împreună cu TQ şi Tr")
Date paraclinice • APTT (TC, TH, PTT) este prelungit *împreună cu TQ şi Tr normale realizează profilul screening comun hemofiliei A, B şi bolii von Willebrand • TGT (timp de generare a tromboplastinei) precizează factorul VIII deficient • Dozarea imunologică a F VIII permite evaluarea cantitativă a nivelului bazal al F VIII 41

Tratamentul HA Progresele în asistenţa bolnavilor de hemofilie au la bază trei “piloni”: • Substituţia F VIII indispensabilă prevenirii şi opririi hemoragiilor • “Comprehensive care” îngrijirea complexă a pacienţilor de echipe specializate care cuprind hematologi, geneticieni, dentişti, ortopezi, nurse, psihologi, asistenţi sociali ş. a. • „Home care” instruirea pacientului sau a persoanei care îl însoţeşte permanent asupra indicaţiei şi modului de administrare profilactică şi terapeutică precoce a concentratelor de F VIII şi acordarea produsului necesar pacientului 42
 Preparate • P-F VIII • RHu-F VIII • PPC *Principalele")
Tratamentul HA (cont. ) Preparate • P-F VIII • RHu-F VIII • PPC *Principalele cerinţe pentru un preparat de calitate sunt: gradul de purificare siguranţa virusologică. şi 43

Recomandări pentru tratamentul episoadelor hemoragice în hemofilia A. Cazuri de hemoragie Hemoragii minore: - hemartroze Hemoragii majore: - hematoame profunde - extracţie dentară* - traumatism cranian - hemoragii în cavitatea bucală* - epistaxis Hemoragii care pun viaţa în pericol: - operaţii majore - sângerări gastrointestinale - hemoragii intracraniene Nivel plasmatic necesar de F VIII Durata 30% Minim 1 zi 40 -50% 3 -4 zile 60 -100% 7 zile , apoi 30 -50% înca 7 zile 44
 Pacienţii care prezintă inhibitori: • Megadoze de F VIII •")
Tratamentul HA (cont. ) Pacienţii care prezintă inhibitori: • Megadoze de F VIII • F VIII porcin • terapia de inducere a toleranţei imune • F VIIa 45
 Probleme: terapia profilactică cu F VIII 46")
Tratamentul HA (cont. ) Probleme: terapia profilactică cu F VIII 46

HEMOFILIA B • este o coagulopatie ereditară Xlinkată datorată deficitului de F IX. • gena F IX este situată la nivelul braţului lung al cromozomului X • incidenţa este de 10 ori mai mică decât a hemofiliei A. 47
 Ancheta familială, datele clinice şi testele screening sunt identice în")
HEMOFILIA B (cont. ) Ancheta familială, datele clinice şi testele screening sunt identice în hemofilia A şi B. 48

Tratamentul hemofiliei B Principiile terapeutice sunt identice cu hemofilia A. *Hemofilia B necesită substituţia F IX şi deci tratamentul se face cu preparate specifice. 49
 Preparate: • P-F IX • concentratele de complex protrombinic")
Tratamentul hemofiliei B (cont. ) Preparate: • P-F IX • concentratele de complex protrombinic (F II, VII, IX, X) • PPC 50
 Inhibitorii FIX au fost semnalaţi extrem de rar iar")
Tratamentul hemofiliei B (cont. ) Inhibitorii FIX au fost semnalaţi extrem de rar iar în aceste cazuri, tratamentul cu FVIIa a dat rezultate bune. 51

BOALA VON WILLEBRAND • Bv. W este o boală genetică determină tulburarea în care anomalia F v. W hemostazei prin deficitul agregării plachetare şi alterarea funcţională a F VIII. • Gena Bv. W este situată la nivelul cromozomului 12 iar transmiterea este autosomal dominantă. • Incidenţa bolii nu este cunoscută exact datorită existenţei formelor fruste dar se estimează că 1% din populaţia de ambele sexe ar fi afectată 52

Fiziopatologia Bv. W Fv. W este o glicoproteină care face parte din complexul macromolecular al FVIII, îndeplinind două funcţii : • proteină transportoare plasmatică a moleculei FVIII. Absenţa Fv. W este urmată de deficit secundar al FVIII prin reducerea sintezei şi creşterea clearance-ului. Efectul este alterarea sistemului intrinsec al coagularii. • controlul agregării trombocitare. Lezarea endoteliului vascular este urmată de creşterea concentraţiei locale a F v. W ce mediază aderarea trombocitelor la endoteliul lezat şi recrutarea de noi trombocite, proces denumit agregare trombocitară. Anomaliile cantitative şi calitative ale F v. W determină un defect calitativ al timpului trombocitar al hemostazei. 53
 Fv. W se comportă ca un reactant de fază")
Fiziopatologia Bv. W (cont. ) Fv. W se comportă ca un reactant de fază acută şi nivelul seric variază în timp: creşte în colagenoze, sarcină, stress chirurgical, infecţii etc. Acest fapt explică: - evoluţia imprevizibilă a cazurilor în timp - dificultăţi de încadrare diagnostică 54

Variantele Bv. W Heterogenitatea clinico-biologică se explică prin expresivitatea variabilă a genei şi prin existenţa a 7 variante de boală: • tip 1, tip 2 A, 2 B, tip trombocitar, 2 N, 2 M şi tip 3 Tipul 1 este cel mai frecvent (70 -80% din cazuri) dar este o formă relativ uşoară şi răspunde la stimularea cu vasopresină. Celelalte tipuri sunt mai rare dar adesea mai severe. 55

Aspecte clinice ale Bv. W Majoritatea cazurilor sunt de gravitate redusă şi sunt relevate de: • epistaxis spontan recidivant şi prelungit • echimoze • menoragii • sângerări prelungite după intervenţii chirurgicale 56
 • Formele severe au manifestare similară hemofiliei")
Aspecte clinice ale Bv. W (cont. ) • Formele severe au manifestare similară hemofiliei dar hemartrozele sunt rare. • Caracterul ambele familial sexe şi apariţia reprezintă la indicii 57 preţioase pentru suspiciunea clinică de

Diagnosticul bolii von Willebrand • TS prelungit la un pacient fără trombocitopenie • APTT (TC, TH) prelungit • dozarea imunologică a Fv. W (scăzut în toate tipurile) • dozarea imunologică a FVIII (scăzut în tip 1, 2 N şi 3) • agregarea trombocitară la ristocetină (deficitară în tip 1, 2 A, 2 M şi 3) 58

Tratamentul bv. W Principiile de terapie sunt aceleaşi pentru bv. W si hemofilie. Preparate: • P-Fv. W • P-F VIII/Fv. W * F VIII recombinant şi concentratele înalt purificate de FVIII nu conţin F v. W şi deci sunt ineficiente . 59
 • Transfuziile de trombocite în tipul trombocitar • DDAVP")
Tratamentul bv. W (cont. ) • Transfuziile de trombocite în tipul trombocitar • DDAVP (desmopresin) elecţie in tipul 1. *Doza este de 0, 3 microg/kg în perfuzie de 20 -30 de minute , la interval de 12 -24 de ore. Există un preparat care se poate administra intranazal (Stimate) în doza de 150 -300 microg. Trebuie menţionat că preparatele de vasopresină utilizate pentru tratamentul 60 diabetului insipid sunt ineficiente datorită dozajului foarte redus.

DEFICITUL DE VITAMINA K Vitamina K este liposolubilă, cu rol în sinteza unor factori coagulanţi (F II, VII, IX, X) şi a unor factori inhibitori (Proteina C, S) Surse: • K 1 – majoritatea alimentelor vegetale 61

Cauzele deficitului de vitamina K • Malabsorbţia – boala celiacă - mucoviscidoza - atrezia biliară - hepatită - enterocolită • Antibioterapia orală prelungită • Deficitul tranzitor de vitamina K al nounăscutului (boala hemoragică a nn) 62

Boala hemoragică, hemoragie cerebrală
 (F VII, X, II) • APTT")
Diagnosticul deficitului de vitamina K • PT (TQ) (F VII, X, II) • APTT (TH, TC) (F IX, X, II) • F II, VII, IX, X 64

Tratamentul deficitului de vitamina K • VITAMINA K 2 -10 mg/zi, 1 -3 zile, iv, sc, po • PPC • Concentrat liofilizat de comlex protrombinic 65

Administrarea profilactică de vitamina K • Nou-născuţi • Alimentaţie parenterală • Antibioterapie prelungită • Medicaţie anticonvulsivantă 66
- Slides: 66