Purine and pyrimidine metabolism Dietary source Purines and




Purine and pyrimidine metabolism Dietary source: • Purines and pyrimidine present in the form of nucleic acid which is present in liver and meat, small amount in legumes, cereals and vegetables. Egg, milk and cheese are not contain nucleic acids. • Tea, coffee, cocoa, and cola contain the methyl purines, caffeine, theobromine and theophylline.

Digestion and absorption: 1 -Pancreas: • Pancreatic juice contains nucleases both ribonucleases and deoxyribonucleases (Rnase and Dnase), that hydrolyse nucleic acids into mononucleotides. 2 -Intestine: • Nucleotidases and intestinal phosphatases remove phosphate producing nucleotides. • Nucleosidases hydrolyse nucleosides into free bases (purine or pyrimidine), and pentose (ribose or deoxyribose). • Nucleotides and nucleosides are poorlyabsorbed from the small intestine, if absorbed-they are catabolized by the liver.

• Although the humans ingest larg amounts of nucleotides and bases they are not utilized for nucleic acid synthesis. • Humans-and other vertebrates- can synthesized ample amounts of purine and pyrimidine nucleotides de novo. • So, purine and pyrimidine bases are not essential dietary components. • Unabsorbed purines are converted to uric acid by intestinal bacteria which may be absorbed and subsequently excreted in urine.

Purine metrabolism Biosynthesis: There are two pathways for purine biosynthesis: 1 -De novo pathway. 2 -Purinessalvage pathway.

1 -De novo pathway • Most of tissues are capable of synthesizing their own purines de novo. • The biosynthetic origins of purine ring atoms:
. Ribose-5 -phosphate is obtained from")
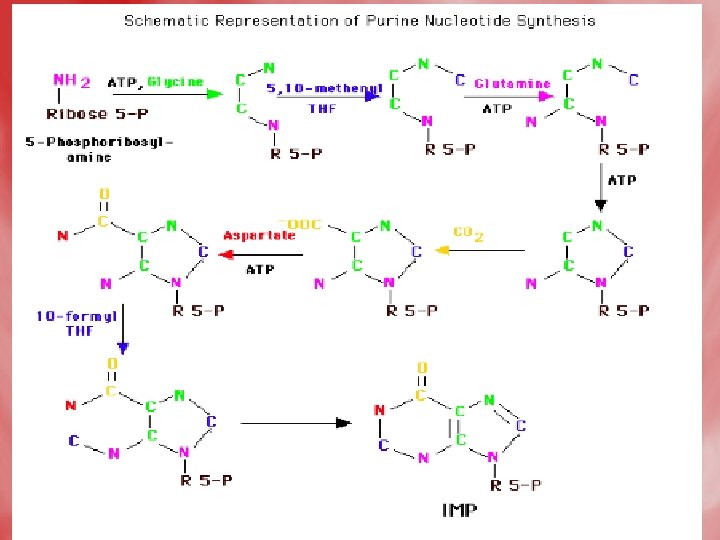
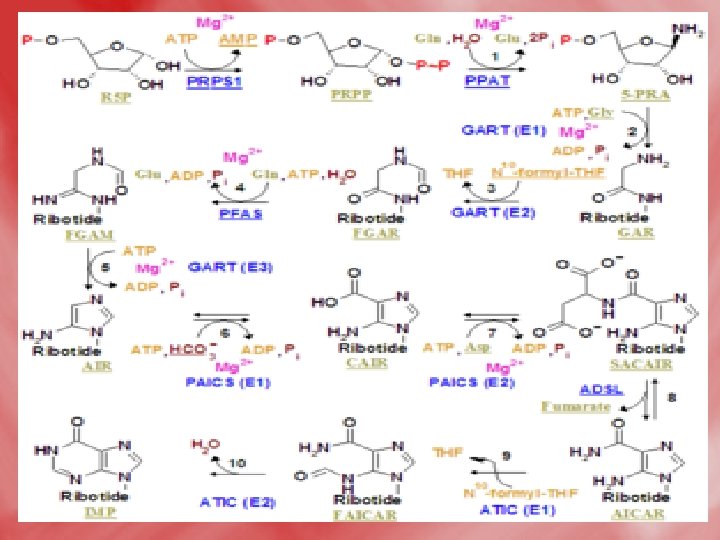
Steps • Step 1: Formation of 5 -phosphoribosyle-1 pyrophosphate(PRPP). Ribose-5 -phosphate is obtained from HMP shunt. This reaction is catalyzed by PRPP synthetase. • Step 2: PRPP is converted into 5 -phosphoribosylamine, by an amidotransferase enzyme, using the amide nitrogen of glutamine as a doner of amino group.

• Step 3: Series of reactions occurs, where the purine ring is built up on the amino group of phosphoribosylaminse, which becomes N-9 of purine. • The product is inosine monophosphate (IMP). • Synthesis of the first fully formed purine nucleotide, inosine monophosphate, IMP begins with 5 -phospho-α-ribosyl-1 pyrophosphate, PRPP. Through a series of reactions utilizing ATP, tetrahydrofolate (THF) derivatives, glutamine, glycine and aspartate this pathway yields IMP. The rate limiting reaction is catalyzed by glutamine PRPP
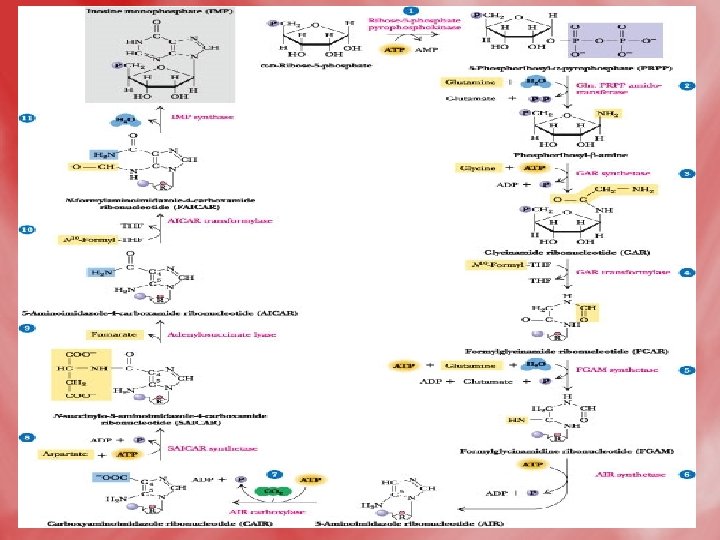

• Enzyme names: 1. glutamine phosphoribosylpyrophosphate amidotransferase 2. glycinamide ribotide synthase 3. glycinamide ribotide transformylase 4. formylglycinamide synthase 5. aminoimidazole ribotide synthase 6. aminoimidazole ribotide carboxylase 7. succinylaminoimidazolecarboxam ide ribotide synthase 8. adenylosuccinate lyase 9. aminoimidazole carboxamide ribotide transformylase 10. IMP cyclohydrolase


• Step 4: Synthesis of AMP and GMP from IMP • IMP is converted to AMP by accepting amino group from aspartate. GTP is hydrolyzed to GDP and Pi. • IMP may be oxidized by IMP dehydrogenase into XMP which is converted to GMP by accepting amino group from glutamine. This reaction needs ATP.
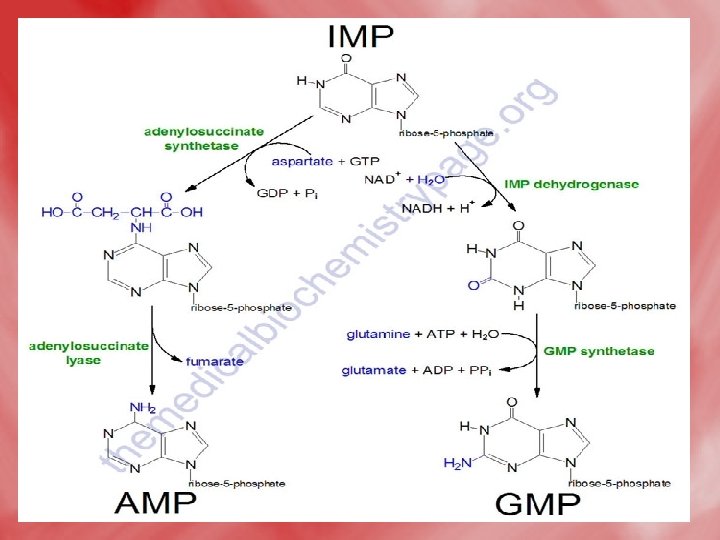
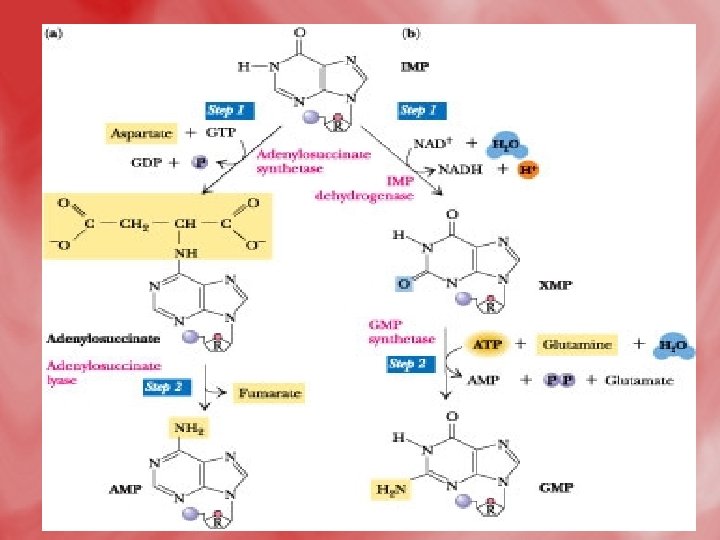

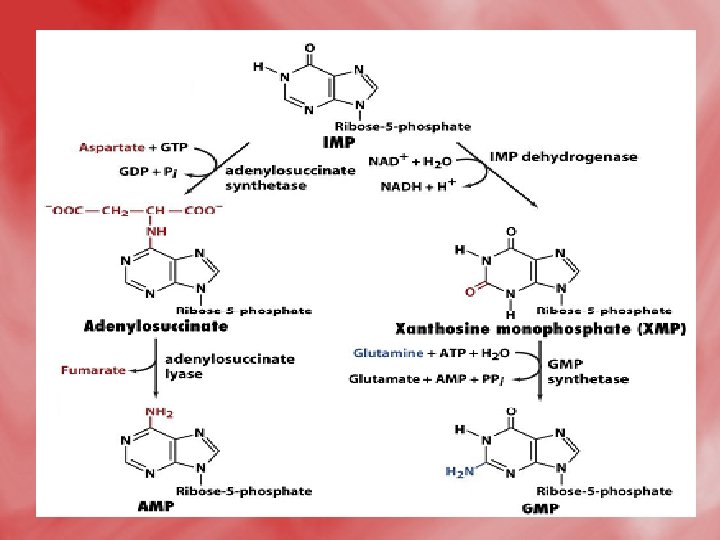

2 -Salvage pathway • A salvage pathway is a pathway in which nucleotides (purine and pyrimidine) are synthesized from intermediates in the degradative pathway for nucleotides. • Salvage pathways are used to recover bases and nucleosides that are formed during degradation of RNA and DNA. This is important in some organs because some tissues cannot undergo de novo synthesis. • The salvaged bases and nucleosides can then be converted back into nucleotides.

Salvage pathway of purine : • This involves conversion of purines resulting from normal turnover of cellular nucleic acids or absorbed from the diet into the corresponding nucleotides. • This saves purines and purine nucleosides from undergoing further catabolism and excretion. It thus decreass de novo synthesis and decreases uric acid production.

There are two mechanisms: 1 -Phosphoribosylation of purines: • Purines from turnover of nucleic acids (or from food) can also be salvaged and reused in new nucleotides. • The enzymeadenine phosphoribosyltransferase (APRT) salvages adenine. • The enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT) salvages guanine and hypoxanthine.

Salvage pathways for purine nucleotides

2 -Phosphoribosylation of purines nucleotides: • By adenosine kinase adenosine converted to adenosine monophosphate. Adenosine kinase • Adenosine AMP ATP ADP
 can also contribute to the salvage")
Purine nucleotide cycle • Purine nucleotide phosphorylases (PNPs) can also contribute to the salvage of the bases through a reversal of the catabolism pathways. However, this pathway is less significant than those catalyzed by the phosphoribosyltransferases. • The synthesis of AMP from IMP and the salvage of IMP via AMP catabolism have the net effect of deaminating aspartate to fumarate. This process has been termed the purine nucleotide cycle. This cycle is very important in muscle cells. Increases in muscle activity create a demand for an increase in the TCA cycle, in order to generate more NADH for the production of ATP. However, muscle lacks most of the enzymes of the major anapleurotic reactions. Muscle replenishes TCA-cycle intermediates in the

• The purine nucleotide cycle serves an important function within exercising muscle. The generation of fumarate provides skeletal muscle with its' only source of anapleurotic substrate for the TCA cycle. In order for continued operation of the cycle during exercise, muscle protein must be utilized to supply the amino nitrogen for the generation of aspartate. The generation of asparate occurs by the standard transamination reactions that interconvert amino acids with αketoglutarate to form glutamate and glutamate with oxaloacetate to form aspartate. Myoadenylate deaminase is the muscle-specific isoenzyme of AMP deaminase, and deficiencies in myoadenylate deaminase lead to post-exercise fatigue, cramping and myalgias.

• The purine nucleoside cycle for anaplerotic replenishment of citric acid cycle intermediates in skeletal muscle.

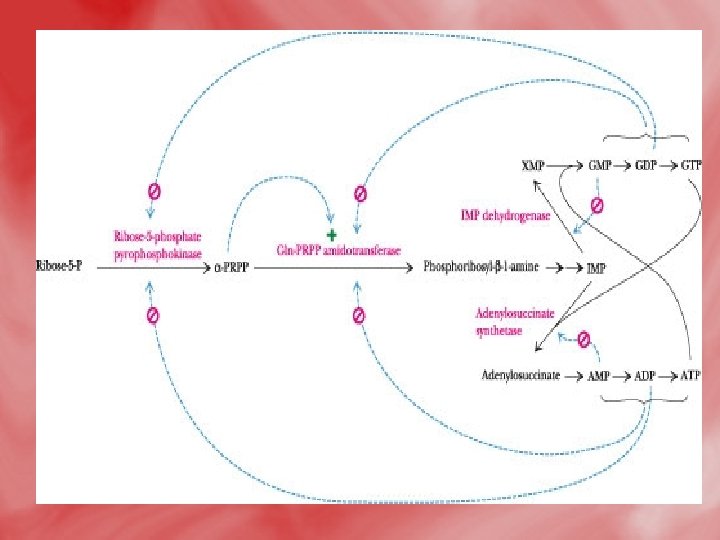
Regulation of purine nucleotide synthesis • The essential rate limiting steps in purine biosynthesis occur at the first two steps of the pathway. The synthesis of PRPP by PRPP synthetase is feed-back inhibited by purine-5'nucleotides (predominantly AMP and GMP). Combinatorial effects of those two nucleotides are greatest, e. g. , inhibition is maximal when the correct concentration of both adenine and guanine nucleotides is achieved. • The amidotransferase reaction catalyzed by PRPP amidotransferase is also feed-back inhibited allosterically by binding ATP, ADP and AMP at one inhibitory site and GTP, GDP and GMP at another. Conversely the activity of the enzyme is stimulated by PRPP. • Additionally, purine biosynthesis is regulated in the branch pathways from IMP to AMP and GMP. The accumulation of excess ATP leads to accelerated synthesis of GMP, and
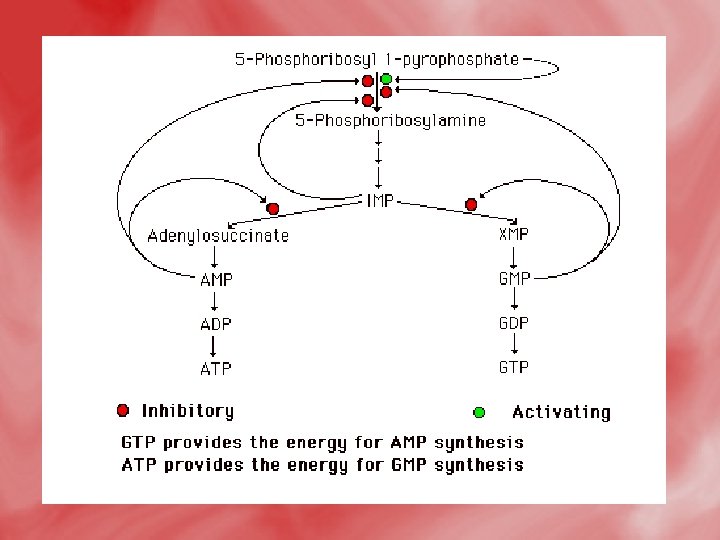
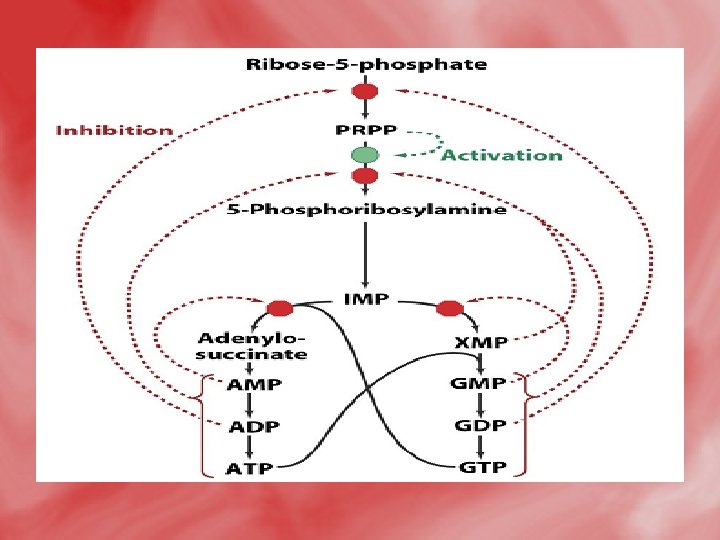

Catabolismof purine 1 -In tissues: • Nucleases hydrolyse nucleic acid forming nucleotides (AMP and GMP). • Nucleotidases act on nucleotide forming phosphates and nucleosides (adenosine, inosine and guanosine).

2 -In liver: • Hypoxanthine is oxidized into xanthine, and further to uric acidby xanthine oxidase. • Uric acid diffuses to the blood to be excreted by the kidney. • N. B. uric acid is the end product of purine base catabolism in main. • In lower mammals uric acid is oxidized into highly water soluble allantion by uricase enzyme. • In mammals (including humans), urea is the end product of protein metabolism (ureotelic). • In birds uric acid is the end product of protein catabolism (uricotelic). • In fishes ammonia is the end protuct of protein catabolism (ammonotelic).
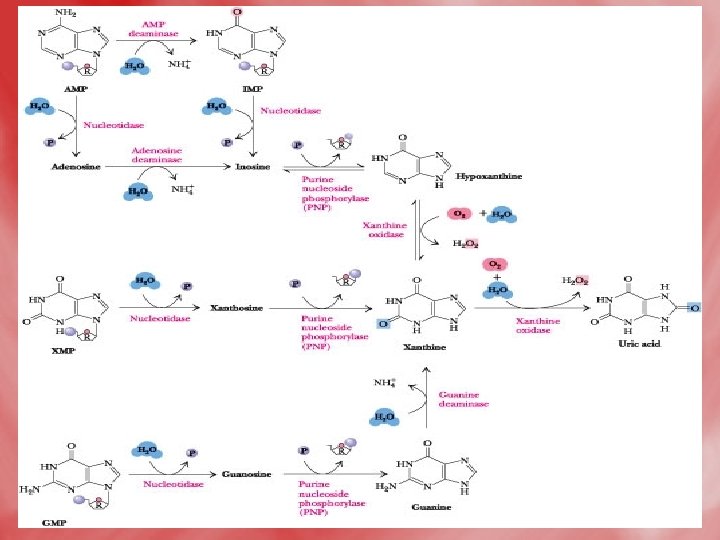
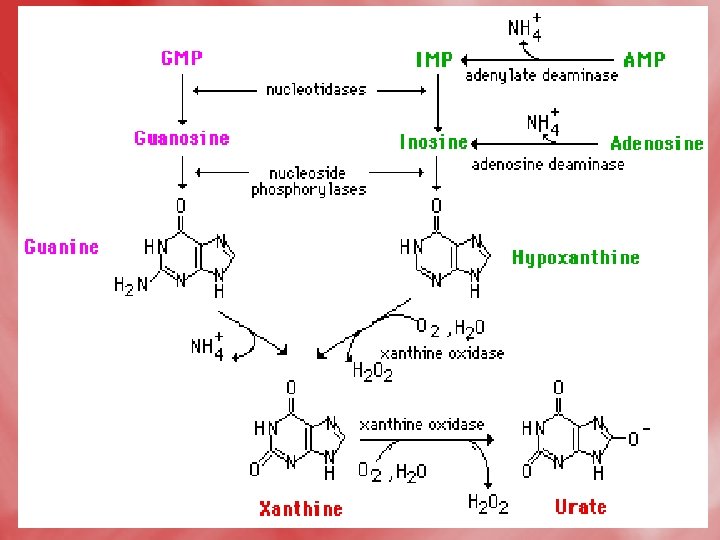
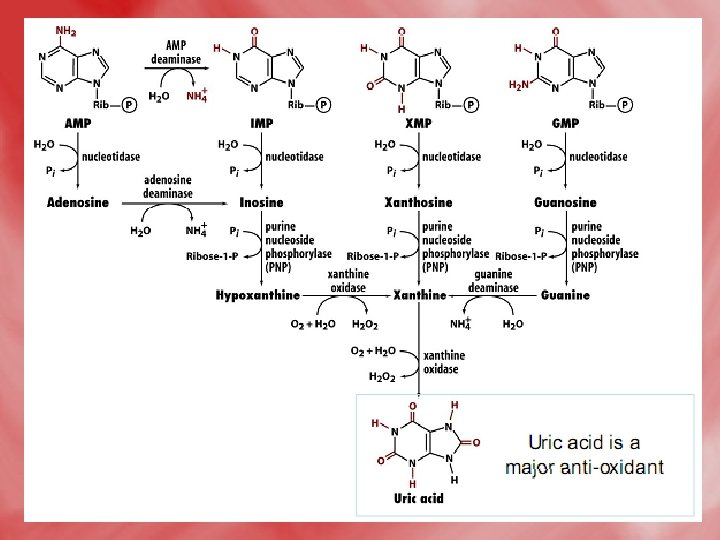
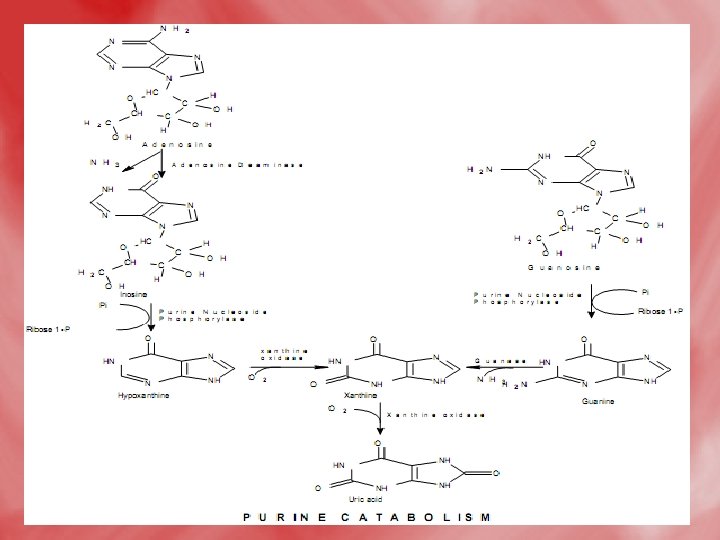

• The Fate of Uric Acid

Overview


Gout • Gout is a metabolic disorder that is related to excess production and deposition of uric acid crystals (hyperuricemia). Uric acid is the byproduct of purine nucleotide catabolism. hyperuricemia is characterized by recurrent attacks of acute inflammatory arthritis. The formation of urate crystals leads to the formation of tophaceous deposits (sandy, gritty, nodular masses of urate crystals (Sodium urate crystals ), particularly in the joints which precipitates the episodes of gouty arthritis. Gouty arthritis is the most painful manifestation of gout and is caused when urate crystals interact with neutrophils triggering an inflammatory response. • Acute gouty arithritic can progress to chronic gouty arithritis and might develop renal stones composed mainly of uric acid and /or urates. • Normall blood uric acid is 3 -7 mg/dl (males) and 2 -6 mg/dl (females).
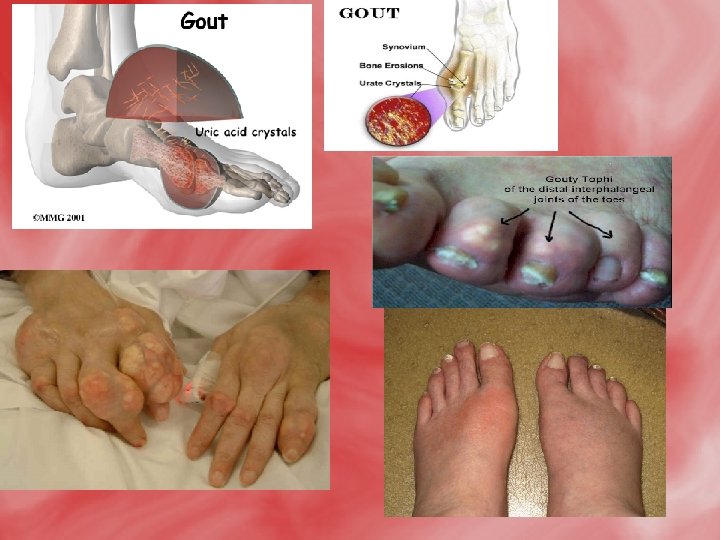

Causes of gout: • Gout may be either metabolic or renal. 1 -The metabolic causes of gout: a-Primary metabolic. b-Secondary metabolic. 2 -The renal causes of gout: a-Primary renal. b-Secondary renal.

Metabolic causes: A-Primary metabolic: • This is due to increased de novo synthesis of purines, that is usually inherited metabolic defect. • In most cases are due to icreased activity of PRPPsynthetase with consequent purine over production. • Deffeciency the glucose-6 -phosphatase enzyme (Von -Gierke´s disease) leads to shunt of glucose-6 phosphate to HMP shunt with final formation of ribose -5 -phosphateand increased intracellular level of PRPP which is positive effector of amido transferase. • Lesch-Nyhan syndrom.

Lesch-Nyhan Syndrome • Lesch-Nyhan syndrome (also known as Nyhan’s syndrome, Kelley-Seegmiller syndrome and Juvenile gout ) is inherited as an X-linked recessive disorder. Since it is an X-linked disease it is found almost exclusively in males although affected females have been identified albeit very rarely. It is a disorder related to defects in the activity of the purine nucleotide salvage enzyme, hypoxanthine-guanine phosphoribosyltransferase (HGPRT).
, produced by")
Causes: • Caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT), produced by mutations in the HPRT gene. The lack of HGPRT causes a build-up of uric acid in all body fluids: both hyperuricemia and hyperuricosuria, which lead to problems such as severe gout and kidney problems, poor muscle control, and moderate mental retardation. These complications usually appear in the first year of life.

• The metabolic consequences of congenital HGPRT deficiency in Lesch. Nyhan syndrome. Loss of HGPRT leads to elevated PRPP levels and stimulation of de novo purine synthesis. One ultimate consequence is increased production of uric acid.

There are three over-lapping clinical syndromes associated with deficiencies in HGPRT activity: • Hyperuricemia. • Neurological proplems e. g. spasticity and mental retardation. • Self mutation.

Features and Characteristics The following characteristics have been identified in individuals with LNS: • Hyperuricemia (overproduction of uric acid) • Urate crystal formation (orange, crystal-like deposits found in the urine, caused by the overproduction of uric acid) • Mental retardation (typically in the moderate range) • Aggressive and impulsive behaviors (always including selfinjurious behaviors) • Choreoathetosis (involuntary writhing movements of the arms and legs and purposeless repetitive movements) • Spasticity
 Ballismus (violent flinging movements")
• • • Dystonia (involuntary spasms and muscle contractions) Ballismus (violent flinging movements of the limbs) Impaired kidney function Irritability Muscle weakness (hypotonia) Speech impairment Hyperrefelxia (exaggeration of reflexes) Kidney stones Blood in the urine Pain and swelling in the joints Difficulty swallowing and eating Vomiting
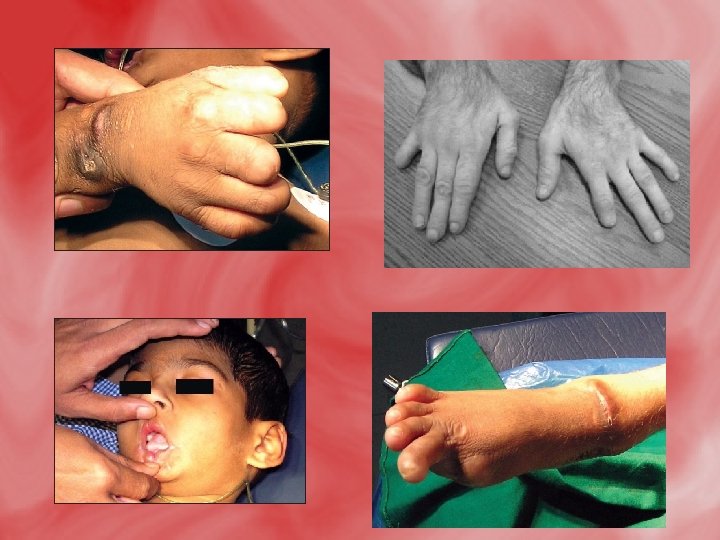

Diagnosis • The overproduction of uric acid is often evident in urine studies and uric acid levels in the blood are typically elevated. However, there are many different causes of hyperuricemia (other than LNS) and some patients with LNS actually have serum uric acid levels that fall into the normal range. Therefore, the detection of hyperuricemia in the blood or urine does not provide reliable diagnostic information. Therefore, a definitive diagnosis of LNS can be obtained by the measurement of the HPRT enzyme in the blood or tissue, or by determining a molecular genetic mutation in the HPRT gene.

2 -Renal causes: A-Primary renal: • Due to inherited defect in the kidney which characterized by decreased tubular secretion of uric acid (decreased renal clearance). B-Seconary renal : • As in: chronic renal failure, due to decreased glomular filteration rate (GFR) and uric acid execrtion. • Lead poisoning that significantly damages the kidney.

Hypouricemia: • It may be renal or metabolic. 1 -renal: due to low renal threshold. 2 -Metabolic: there is decreased uric acid formation due to deficiency of: A-adinosine deaminase, (adenosinuria). b-Xanthine oxidase, hypoxanthine and xanthine are excreted in excess in urine. Xanthine stone might be formed. C- purine nucleosidase. there is accumulation of guanosine and inosine leading to guanosinuria. D-Folic acid. There is decreased de novo synthesis of purines.

Symptoms • In addition to the symptoms associated with immunodeficiency, such as depletion of T-cells, decline of lymphocyte activity, and an abrupt proliferation of both benign and opportunistic infections, PNP-deficiency is often characterized by the development of autoimmune disorders. Lupuserythematosis, autoimmune hemolytic anemia, and idiopathic thrombocytopenic purpura have been reported with PNP-deficiency. • Neurological symptoms, such as developmental decline, hypotonia, and mental retardation have also

Pyrimidine nucleotides metabolism • The ring of pyrimidine is synthesized before being attached to ribose-5 -phosphate which is donated by PRPP. • The source of carbon and nitrogen atoms in the pyrimidine ring as follow:

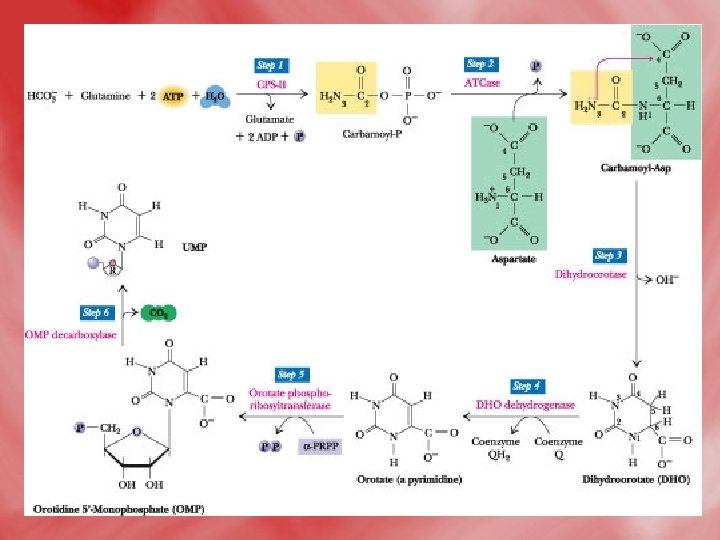
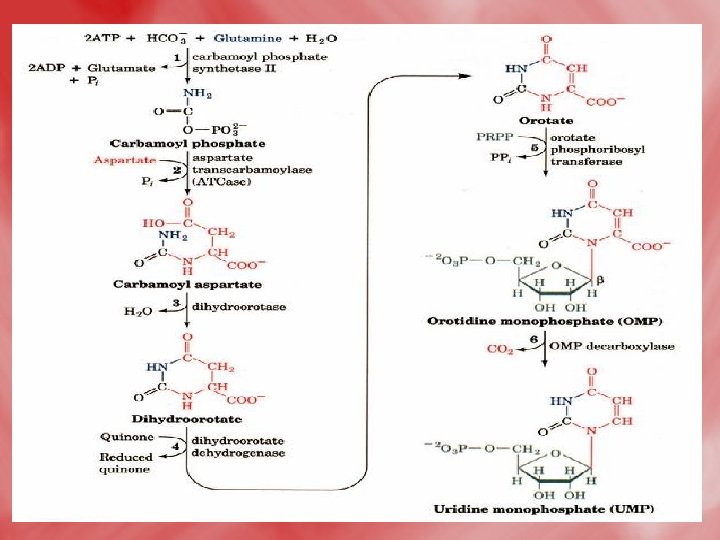
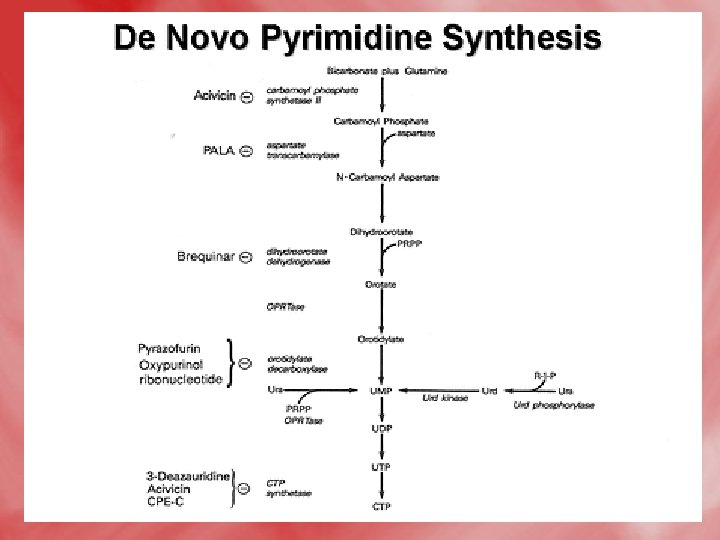
1 -De novo synthesis of pyrimidines: • All reactions occure in the cytosol except for dihydroorotate dehydrogenase reaction which is mitochondrial (inner mitochondrial membrane). 1 -formation of carbamoyle phosphate, and the reaction is catalsed by carbamoyl phosphate synthetase II. • N. B. this enzyme is distinct from carbamoyl phosphate synthetase I of urea cycle, which is a mitochondrial enzyme, on the other hand, the α–amide group of glutamine acts as nitrogen donner (NH 3), for carbamoyl phosphate synthetase II, unlike ammonia (NH 3+)that donates nitrogen for carbamoylphosphate synthetase. I.
. Note that,")
• The reaction catalyzed by carbamoyl phosphate synthetase II (CPS II). Note that, in contrast to carbamoyl phosphate synthetase I, CPS II uses the amide of glutamine, not NH 4+, to form carbamoyl-P. Step 1: The first ATP consumed in carbamoyl phosphate synthesis is used in forming carboxyphosphate, an activated form of CO 2. Step 2: Carboxyphosphate (also called carbonyl-phosphate) then reacts with the glutamine amide to yield carbamate and glutamate. Step 3: Carbamate is phosphorylated by the second ATP to give ADP and carbamoyl phosphate.

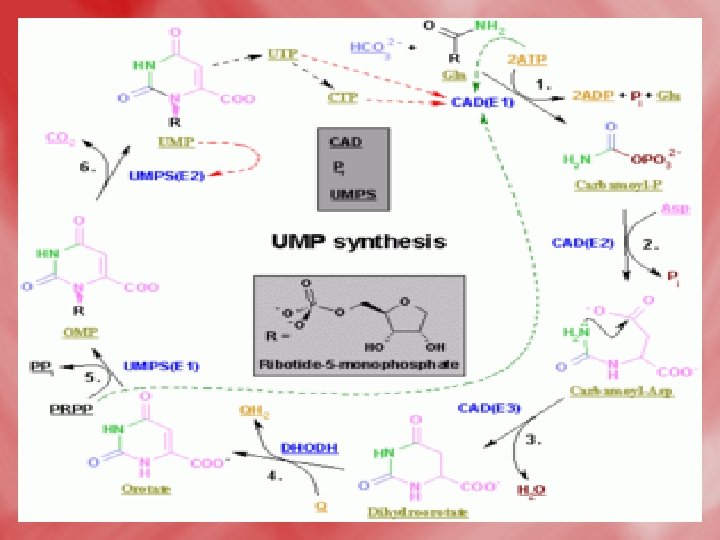
2 -Condensation of carbamoyl phosphate with aspartate forming carbamoyl aspartate. 3 -ring closure, associated with H 2 O loss forms dihydroorotic acid. 4 -orotic acid is formed from dihydroorotate by removal of hydrogen from C 5 and C 6. 5 -Transfere of ribose phosphate from PRPPto form orotidine monophosphate (OMP). 6 -Decarboxylation to form uridine monophosphate (UMP). UTP is formed from UMP by receiving phosphates from ATP.

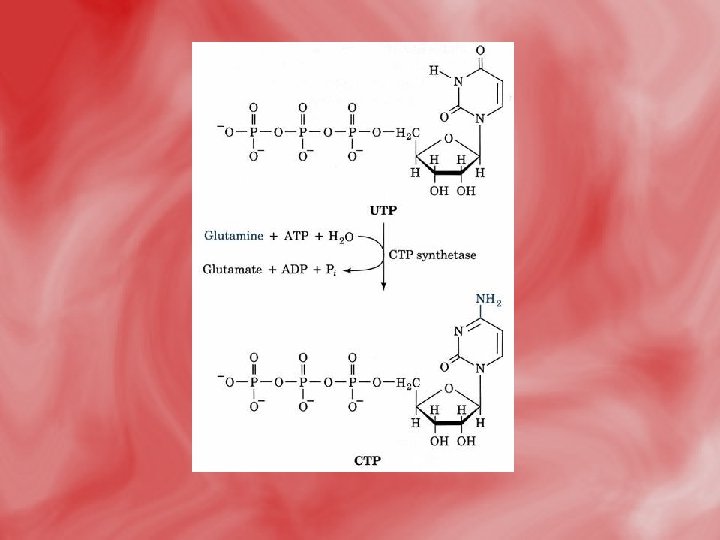
7 -UTP takes amino group from glutamine to form CTP. UTPand CTP can be utilized for RNA synthesis. 8 -the conversion of ribose to deoxyribose occurs while in the form of nucleoside diphosphate, and is catalysed by ribonucleotide reductase that needs thioredoxin ( which is a small protein that contain two sulfhydryle SH groups, hydrogen donner). • N. B> the cellular level of deoxyribonucleotides is usually low. It is increased only at time of DNA replication, since all nucleotides are synthesized originally as ribonucleotides. 9 -The methyl gropup of thymidylate (d. TMP) is derived from methyelene FH 4, which is converted to dihydrofolate in a reaction catalyzed bythymidylate synthase.
 • Step")
The de novo pyrimidine biosynthetic pathway. (by another way to increase understanding) • Step 1: Carbamoyl-P synthesis. • Step 2: Condensation of carbamoyl phosphate and aspartate to yield carbamoyl-aspartate is catalyzed by aspartate transcarbamoylase (ATCase). • Step 3: An intramolecular condensation catalyzed by dihydroorotase gives the six-membered heterocyclic ring characteristic of pyrimidines. The product is dihydroorotate (DHO). • Step 4: The oxidation of DHO by dihydroorotate dehydrogenase gives orotate. (In bacteria, NAD+ is the electron acceptor from DHO. ) • Step 5: PRPP provides the ribose-5 -P moiety that transforms orotate into orotidine-5'-monophosphate, a pyrimidine nucleotide. Note that orotate phosphoribosyltransferase joins N-1 of the pyrimidine to the ribosyl group in appropriate b-configuration. PPi hydrolysis renders this reaction thermodynamically favorable. • Step 6: Decarboxylation of OMP by OMP decarboxylase yields UMP.

• CTP synthesis from UTP. CTP synthetase catalyzes amination of the 4 -position of the UTP pyrimidine ring, yielding CTP. In eukaryotes, this NH 2 comes from the amide-N of glutamine; in bacteria, NH 4+ serves this role.

Enzyme names: 1. aspartate transcarbamoylase, ATCase 2. carbamoyl aspartate dehydratase 3. dihydroorotate dehydrogenase 4. orotate phosphoribosyltransferase 5. orotidine-5'-phosphate carboxylase

Synthesis of d. TMP from d. UMP

Pyrimidine Synthesis


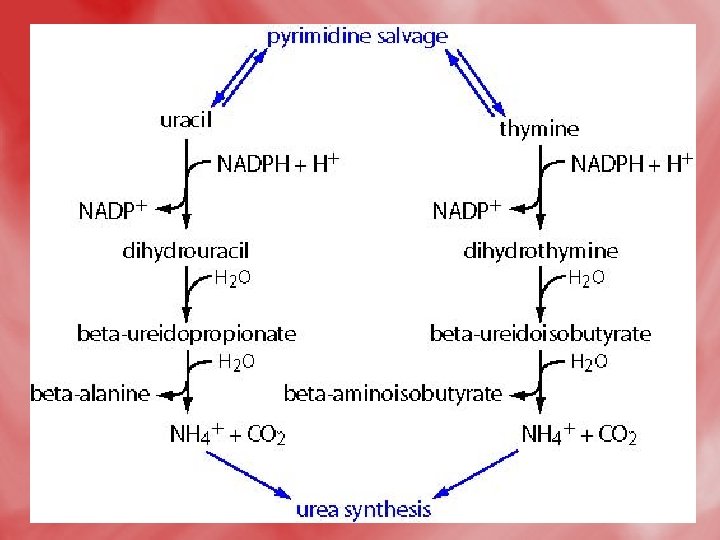
Catabolism of pyrimidines: • This occursmainly in the liver. • β -alanine is the major end product of both uracil and cytosine. • β–aminoisobutyric is the major end product of thymine. • The release of respiratory CO 2 from pyrimidine nucleus (C 2) occurs in the course of catabolism of three pyrimidine bases. • The reactions as follow:
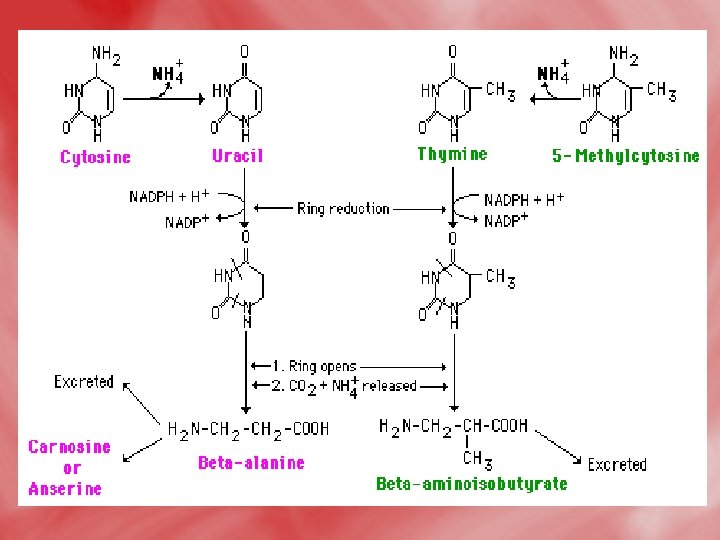

• Pyrimidine degradation. Carbons 4, 5, and 6 plus N-1 are released as β alanine, N-3 as NH 4+, and C-2 as CO 2. (The pyrimidine thymine yields β aminoisobutyric acid. ) Recall that aspartate was the source of N-1 and C 4, -5, and -6, while C-2 came from CO 2 and N-3 from NH 4+ via glutamine.

Disorder of pyrimidine metabolism 1 -β-amino isobutyric aciduria: • This characterized byexecretion of larg amounts of βaminoisobutyric in urine. • Causes: • This is the result of defective deamination of βaminoisobutyric acid. • The condition also occures in cases of leukemia, and exposure to radiation due to increased DNA breakdown.

2 -Hereditary orotic aciduria: Disorder aciduria, Type I Defective Enzyme Comments orotate phosphoribosyl transferase and OMP decarboxylase Orotic aciduria, Type II OMP decarboxylase causes retarded growth, and severe anemia caused by hypochromic erythrocytes and megaloblastic bone marrow. Leukopenia is also common in orotic acidurias. As type 1 • The disorders can be treated with uridine and/or cytidine, which leads to increased UMP production via the action of nucleoside kinases. The UMP then inhibits CPS-II, thus attenuating orotic acid production.
 the urea cycle enzyme, ornithine")
Orotic aciduria due to OTC deficiency (no hematologic component) the urea cycle enzyme, ornithine transcarbamoylas e, is deficient increased mitochondrial carbamoyl phosphate exits and augments pyrimidine biosynthesis; hepatic encephalopathy drug induced orotic aciduria OMP decarboxylase allopurinol and 6 azauridine treatments cause orotic acidurias without a hematologic component; their catabolic by-products inhibit OMP


• http: //images. google. com. eg/imgres? imgurl=http: //w ww. goutaware. com/images/De. Novo. Synthesis. Purine. Nucleotid es. gif&imgrefurl=http: //www. goutaware. com/Synthesis-of-Purine. Nucleotides. html&usg=__bndrw 0 so. Py 8 F 9 o. IRd 6 tt. Rcf. Gq. A=&h=787&w=600&sz=4&hl= ar&start=23&tbnid=p. Pm. Ff. As. Xta 1 FM: &tbnh=143&tbnw=109&prev=/images% 3 Fq%3 DNucleotides%26 gbv%3 D 2%26 ndsp%3 D 20% 26 hl%3 Dar%26 sa%3 DN%26 start%3 D 20 • http: //themedicalbiochemistrypage. org/nucleotidemetabolism. html • http: //www. macaulay. ac. uk/IFRU/iaeacd/html/techd oc/html/images/26. gif

• http: //images. google. com. eg/imgres? imgurl=http: //www. marefa. org/images/thumb/6/6 b/Nucleotides_syn 2. png/300 px. Nucleotides_syn 2. png&imgrefurl=http: //www. marefa. org/index. php/%25 D 9%2586%25 D 9%258 A%25 D 9%2588%25 D 9%2583% 25 D 9%2584%25 D 9%258 A%25 D 9%2588%25 D 8%25 AA%25 D 8 %25 A 7%25 D 9%258 A%25 D 8%25 AF&usg=__j 9 Lcxv 731 Den. Hg. Si Gt. VQS 9 Ggs. Fg=&h=296&w=300&sz=51&hl=ar&start=18&tbnid= C 8 YJ 4 FRm. SUQe. BM: &tbnh=114&tbnw=116&prev=/images%3 Fq %3 DNucleotides%26 gbv%3 D 2%26 hl%3 Dar%26 sa%3 DG • http: //www. biologylessons. sdsu. edu/ta/classes/lab 6/nucleosides. gif • https: //www. msu. edu/course/isb/202/ebertmay/2004/drivers/n ucleotide. jpg • http: //upload. wikimedia. org/wikipedia/commons/b/b 9/Nucleot ides. png • http: //themedicalbiochemistrypage. org/lesch-nyhan. html • http: //themedicalbiochemistrypage. org/gout. html • http: //themedicalbiochemistrypage. org/scid. html • http: //www. neurology. org/cgi/content/full/65/11/E 25/F 151 • http: //themedicalbiochemistrypage. org/scid. html

• http: //images. google. com. eg/imgres? imgurl=http: //web. virgin ia. edu/Heidi/chapter 27/Images/8883 n 27_17. jpg&imgrefurl=h ttp: //web. virginia. edu/Heidi/chapter 27/chp 27. htm&usg=__1 z 28 Mec. AEL 4 v. L 93 Ky. YLzp. Pjiy. M=&h=362&w=508&sz=31&hl=ar&start=50&um =1&itbs=1&tbnid=vr. MSXp. Osm. OOmo. M: &tbnh=93&tbnw=131 &prev=/images%3 Fq%3 Dpyrimidine%2 Bmetabolism%26 ndsp %3 D 20%26 hl%3 Dar%26 sa%3 DN%26 start%3 D 40%26 um%3 D 1 • http: //images. google. com. eg/imgres? imgurl=http: //www. speci alchild. com/archives/posterchild 061. jpg&imgrefurl=http: //www. specialchild. com/archives /dz-031. html&usg=__o. U 4 Xw 6 Fte 79 bp. X 58 pg. Vdj. Rp. CQc=&h=224&w=169&sz=18&hl=ar&start=1&tbnid=0_v_0 ws. Z 5 Qv. DOM: &tbnh=108&tbnw=81&prev=/images%3 Fq%3 DLesch. Nyhan%2 BSyndrome%26 gbv%3 D 2%26 hl%3 Dar%26 sa%3 DG

- Slides: 84