PSI e PHI BLAST Eduardo Sampaio Rocha BLAST

PSI e PHI BLAST Eduardo Sampaio Rocha

BLAST • Basic Local Alignment Search Tool – Desenvolvido por Altschul, Gish, Miller, Myers e Lipman em 1990 – Conjunto de ferramentas web (http: //www. ncbi. nlm. nih. gov/BLAST/) ou de linha de commando. – Em 1997 foi lançada a versão 2. 0 que é 3 x mais rápida que a original

BLAST e Proteínas • BLAST não é muito sensível as similaridades fracas. Estas similaridades podem ser biologicamente importantes. • As famílias de proteínas geralmente são caracterizadas por padrões de regiões conservadas. O BLAST original não permitia a consulta de padrões de proteínas Os dois problemas acima são resolvidos respectivamente por duas ferramentas incorporadas ao BLAST 2. 0: – PSI-BLAST (Posicion-Specific Iterated BLAST) – PHI-BLAST (Pattern-HIT initiated BLAST)

PSI-BLAST • Ferramenta iterativa que usa um profile como entrada para aumentar a sensitividade • O profile é gerado automaticamente a partir dos alinhamentos da saída no passo anterior • O profile é usado na geração de uma nova matriz de score • Muito sensível ao conteúdo da base de dados

PHI-BLAST • Integrado ao PSI-BLAST • Pega como entrada um padrão de proteína e uma seqüência • Procura na base por proteínas que casem com o padrão especificado e que tenham similaridade com a seqüência original • Ferramenta em desenvolvimento!

Fluxograma PSSM Seqüência/Padrão BLAST 2. 0 Alinhamentos > limiar Profile

A Ferramenta • Web – Pode ser encontrada em http: //www. ncbi. nlm. nih. gov/BLAST/ • Linha de Comando – Pode ser baixada em: ftp: //ncbi. nlm. nih. gov/blast

Entrada da Ferramenta • Seqüência no formato FASTA – Exemplo: gi|129295|sp|P 01013|OVAX_CHICK GENE X PROTEIN (OVALBUMIN-RELATED) QIKDLLVSSSTDLDTTLVLVNAIYFKGMWKTAFNAEDTREMPFHVTKQESKPVQMMCMNNSFNVATLPAE KMKILELPFASGDLSMLVLLPDEVSDLERIEKTINFEKLTEWTNPNTMEKRRVKVYLPQMKIEEKYNLTS • Seqüência no formato simples (podendo ter espaços e números) – Exemplo: 1 61 121 181 qikdllvsss sfnvatlpae rrvkvylpqm edgiemagst tdldttlvlv kmkilelpfa kieekynlts gviedikhsp naiyfkgmwk sgdlsmlvll vlmalgmtdl eseqfradhp tafnaedtre pdevsdleri fipsanltgi flflikhnpt mpfhvtkqes ektinfeklt ssaeslkisq ntivyfgryw kpvqmmcmnn ewtnpntmek avhgafmels sp
 • Padrão de aminoácido que se deseja procurar – Exemplo:")
Entrada da Ferramenta (PHI-BLAST) • Padrão de aminoácido que se deseja procurar – Exemplo: [RG]-[M]-[X]-[YWF]-5[X]-[A]

Principais Parâmetros de Entrada • Base de Dados de Seqüências Peptídicas – nr, swissprot, pat, yeast, ecoli, pdb, Drosophila genome e month • Matriz de Substituição – PAM 30, PAM 70, BLOSSUM 80, BLOSSUM 62, BLOSSUM 45 • Custo do Gap – Custo de Inclusão e Extensão • Limiar de inclusão de uma seqüência no modelo usado pelo PSI-BLAST para a geração da PSSM usada na próxima iteração
 – Derivada do alinhamento global de")
Matriz de Substituição • PAM (Percent Accepted Mutation) – Derivada do alinhamento global de seqüências bastante relacionadas – O número da matriz (e. g. PAM 120) se refere a distância evolucional. Assim da PAM 30 nós devemos esperar alinhamentos que são mais próximos na evolução do que a PAM 250 – A matriz de maior número são extrapoladas de menores números
 – Mais sensível a alinhamentos locais")
Matriz de Substituição • BLOSSUM (Block Substitution Matrix) – Mais sensível a alinhamentos locais de seqüências relacionadas – O número da matriz (Blossum 62) é relacionada com a mínima porcentagem de identidade dos blocos usados para construir a matriz – quanto maior o número, menor a distância – Geralmente possuem melhor performance na procura de similaridade local do que as PAMs

Saída do Programa • Formato HTML/XML, arquivo texto ou ASN. 1 • Saída pode ser dividida em: – Descrição do programa – Gráfico mostrando os principais alinhamentos – Descrição dos Alinhamentos ordenados pelo menor Evalue (aqui podemos selecionar quais alinhamentos vão ser considerados na próxima iteração) – Alinhamentos – Estatísticas referentes ao programa

Saída do Programa Alinhamento • E-Value – o valor esperado é a probabilidade que o casamento associado seja devido ao acaso (fator randômico). Depende do tamanho da base de dados e da query. • Score(bits) – é a soma dos valores obtidos para o alinhamento de acordo com a matriz de alinhamento. Quanto maior o score, melhor o alinhamento • Identities – porcentagem do casamento exato entre a seqüência fonte e a seqüência da base de dados • Gaps – porcentagem do número de gaps • Positives – porcentagem do casamento exato e de score positivo.

Montando o Profile • No final de cada iteração, o PSI-BLAST mostra quais alinhamentos foram acima do limiar e quais foram abaixo. • Com um checkbox ao lado de cada alinhamento, o PSI-BLAST da a possibilidade de escolha de qual alinhamento deve entrar na construção do profile que ira servir de entrada para a próxima iteração • Símbolos indicam quando uma seqüência foi levada em consideração na iteração anterior e quando uma seqüência foi encontrada nesta iteração

Exemplo • Encontrar parentes distantes da proteína MJ 0577 da Methanococcus jannaschii gi|2501594|sp|Q 57997|Y 577_METJA PROTEIN MJ 0577 MSVMYKKILYPTDFSETAEIALKHVKAFKTLKAEEVILLHVIDEREIKKRDIFSLLLGVAGLNKSVEEFE NELKNKLTEEAKNKMENIKKELEDVGFKVKDIIVVGIPHEEIVKIAEDEGVDIIIMGSHGKTNLKEILLG SVTENVIKKSNKPVLVVKRKNS • Usar a base de dados nr • Usar a matriz BLOSSUM 62 • Usar um limiar de 0. 001
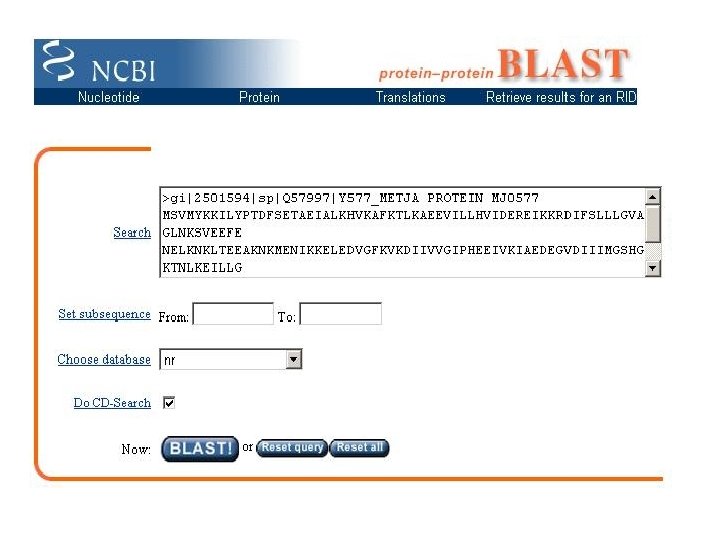


Resultado – Iteração 1

Resultado – Iteração 1




Iteração 2



Referências • http: //www. ncbi. nlm. nih. gov/BLAST/ • Altschul, Stephen F. , Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25: 3389 -3402. • Zheng Zhangm Alejandro A. Schäffer, Webb Miller, Thomas L. Madden, David J. Lipman, Eugene V. Kooning, and Stephen F. Altschul (1998), “Protein sequence similarity searches using patterns as seeds”, Nucleic Acids Res. Vol. 26, No. 17
- Slides: 28