Protein modelling Tutorial Retrieve sequence Automated Mode SWISS






























. Click and choose Target-Template Alignment")





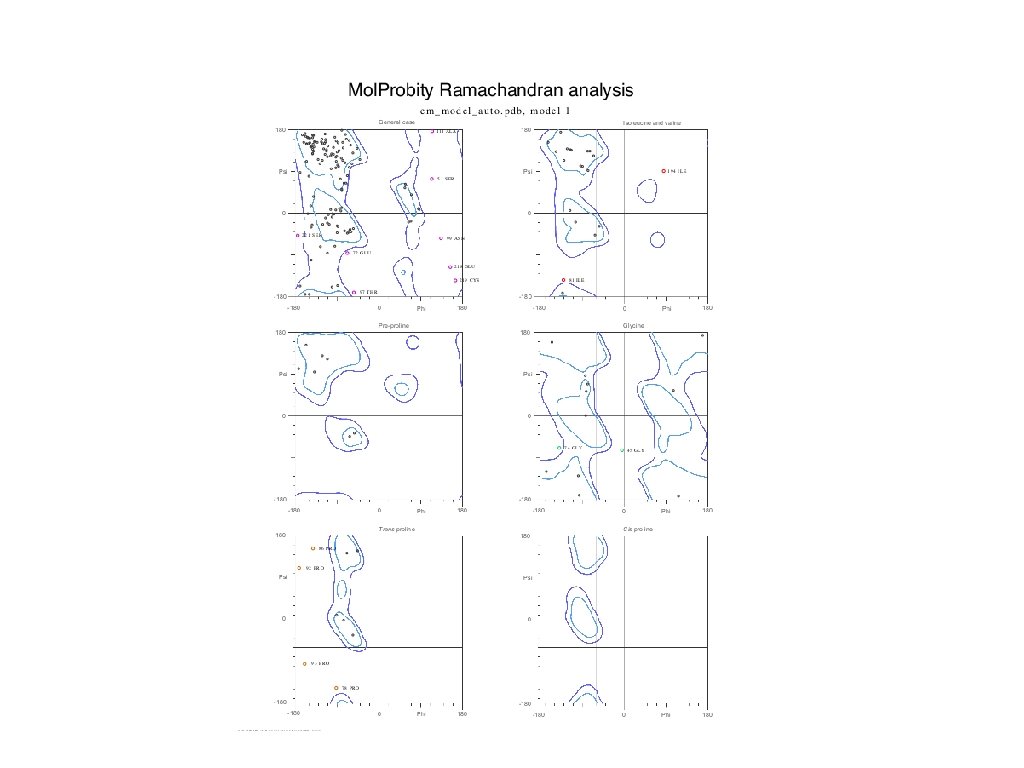
. Download the PDB and submit it to Mol. Probity • Are")





- Slides: 47
Protein modelling Tutorial
Retrieve sequence Automated Mode SWISS MODEL Browser protein information - Uni. Prot find homologous sequences - BLAST template(s) selection multiple sequence alignment - t-coffee template(s) - target alignment Alignment Mode SWISS MODEL prediction Py. Mol tutorial Model assessment Mol. Probity Structural comparison Py. Mol assessment visual analysis
• • modelling the structure of our target protein If you have the sequence of your target protein, and some hits from your template search, your next step is to use an homology modelling software to generate a model of your target protein. SWISS-MODEL (http: //swissmodel. expasy. org) is a webserver for homology modelling. It provides the following two modes of usage: • • Automated mode: Input sequence of target protein. SWISS -MODEL searches for template, performs alignment and then homology modelling for your target protein. Alignment mode: Input sequence alignment of target protein against a template of your choice (searched through e. g. BLAST). SWISS-MODEL models your target protein
Sequence of ARAF kinase domain >query SEVQLLKRIGTGSFGTVFRGRWHGDVAVKVLKVSQPTAEQAQAFKNEMQVLRK TRHVNILLFMGFMTRPGFAIITQWCEGSSLYHHLHVADTRFDMVQLIDVARQTAQGMDYL HAKNIIHRDLKSNNIFLHEGLTVKIGDFGLATVKTRWSGAQPLEQPSGSVLWMAAEVIRM QDPNPYSFQSDVYAYGVVLYELMTGSLPYSHIGCRDQIIFMVGRGYLSPDLSKISSNCPK AMRRLLSDCLKFQREERPLFPQILATI As ARAF contains multiple domains we will restrict our modelling to focus on its kinase domain Adhere to the FASTA format when dealing with Note: sequences: Symbol “>” followed by a title aa sequence in capital letters (beware windows may try to save with an extra. txt extension. This might cause problems!!!)
Create a model of ARAF
Perform automated modelling • Use the ARAF kinase domain sequence we have provided
SWISS-MODEL searches for possible templates Study the information listed here on possible templates – choose (a) template(s) and wait for modelling 7
While the Swiss-Model is running. . . Find homologous structures of the target sequence. The homologous structure(s) you found can be the candidate template to build the predicted structure of your target sequence. Later on, you will have to compare/analyse the template structure you choose and the template chosen by Swiss-Model together with the predicted structure.
sequence from Uni. Prot Automated Mode SWISS MODEL extracted domain from entire sequence Uni. Prot find homologous sequences - BLAST template(s) selection multiple sequence alignment - t-coffee template(s) - target alignment Alignment Mode SWISS MODEL prediction Py. Mol tutorial Model assessment Mol. Probity Structural comparison Py. Mol assessment visual analysis
exercise 1. Use the kinase domain sequence of ARAF we have supplied. 2. Choose the correct blast program to search the content of this entry against the PDB database of protein sequences. 3. What is % of sequence id of the first top 10 hits? 4. Do they cover all the query sequence? 5. Are these top 10 matches statically significant? 6. Which experimental methods were used to determine the structures? 7. Which hit(s) would you choose as a template for modelling the kinase domain of ARAF? 8. Download the fasta sequences of your chosen hits.
blast. ncbi. nlm. nih. gov
Why do we choose the PDB database? blast. ncbi. nlm. nih. gov
Several algorithms are available blast. ncbi. nlm. nih. gov
Understanding the Alignments
Understanding the BLAST Output Graphic Display Hit List Alignments
Understanding the Graphic Display
Understanding the Graphic Display
Understanding the Hit List
Understanding the Alignments
Understanding the Alignments
Understanding the Alignments Low Complexity
Find out more about hit structures on the PDB Put here the PDB identifier and search
Criteria to choose a good template • How good is the alignment? • Coverage? • • Sequence identity? • 30% rule Score? • E-value (this will be used later when we identify templates using BLAST)? • describes the number of hits one can "expect" to see by chance when searching a database of a particular size. The higher it is, the more likely it represents random background noise (more “insignificant”) • Also taken into account the length of the alignments • Ref: read BLAST FAQ • The template • What experimental method was used in determining the structure of the template protein (X-ray crystallography / NMR / Cryo-EM etc. ) • How good is the resolution of the template structure? • Usually, structures solved by X-ray crystallography are preferred for use in homology modelling.
Compare template hits • Compare the table of template hits that SWISS-MODEL and BLAST produces. • Can you observe any differences? • Hits appearing in one but not the other? • Ordering of hits? (How are they ordered? )
sequence from Uni. Prot Automated Mode SWISS MODEL extracted domain from entire sequence Uni. Prot find homologous sequences - BLAST template(s) selection multiple sequence alignment - t-coffee template(s) - target alignment Alignment Mode SWISS MODEL prediction Py. Mol tutorial Model assessment Mol. Probity Structural comparison Py. Mol assessment visual analysis
modelling the structure of our target protein • • If you have the sequence of your target protein, and some hits from your template search, your next steps are: • Perform a sequence alignment of your target against the template. • use an homology modelling software to generate a model of your target protein. SWISS-MODEL (http: //swissmodel. expasy. org) is a web-server for homology modelling. Users input an alignment of their target with a template of choice, and SWISS-MODEL generates the model according to the alignment. (“Alignment mode”).
• Retrieve the template you have chosen earlier on in the BLAST exercise. • Now what we want is to perform a sequence alignment of our target protein against the template. This would be the instruction for SWISS-MODEL to generate a model for our target. • We use T-COFFEE. It is available at: –http: //tcoffee. crg. cat/ –https: //www. ebi. ac. uk/Tools/msa/tcoffee/
T-coffee server for sequence alignment http: //tcoffee. crg. cat/ Click here!
Enter sequences Click here!
Download as fast_aln file
Submit the alignment to SWISS-MODEL (https: //swissmodel. expasy. org). Click and choose Target-Template Alignment on the right: Upload the alignment file from T-Coffee. (Or paste it in this box) Just choose any one.
Analyse models Assess model quality Download model Visualise model
Model quality
save the PDB model structure and submit it to the Mol. Probity server for quality assessment sequence from Uni. Prot Automated Mode SWISS MODEL template(s) selection template(s) - target alignment prediction (Quality Information) assessment Model assessment Mol. Probity Discuss template selection
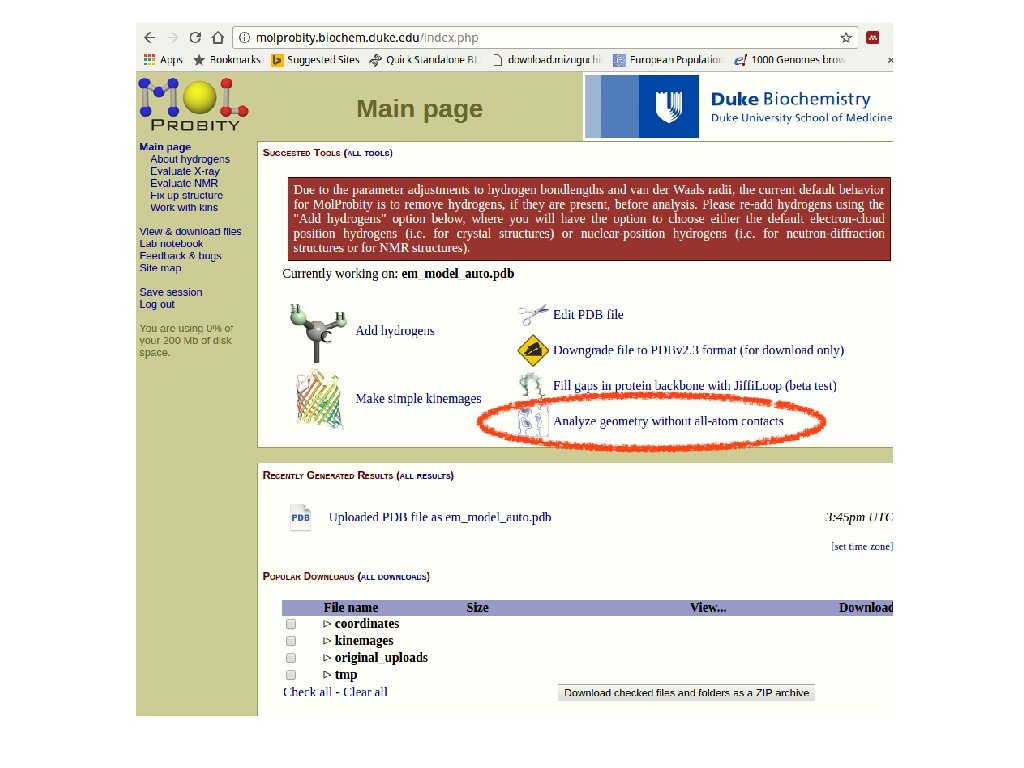
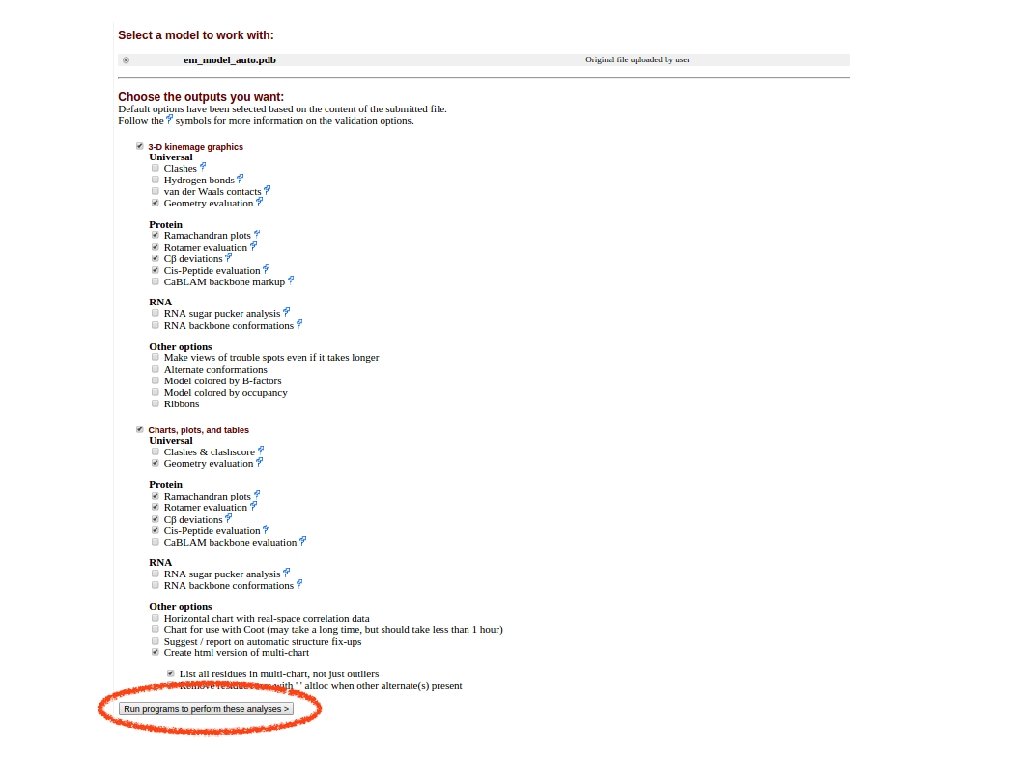
You would want to assess the model! • If you submit the template structure, you would still obtain statistics but this tells you nothing directly about your model! http: //molprobity. biochem. duke. edu
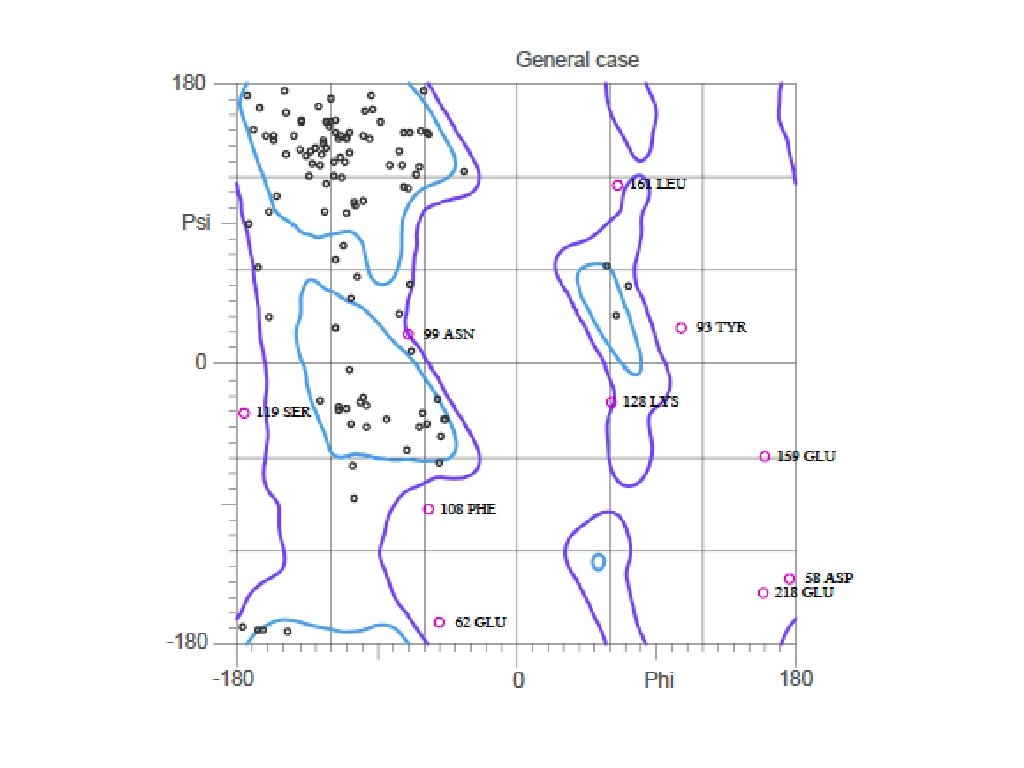
Download Ramachandran plot
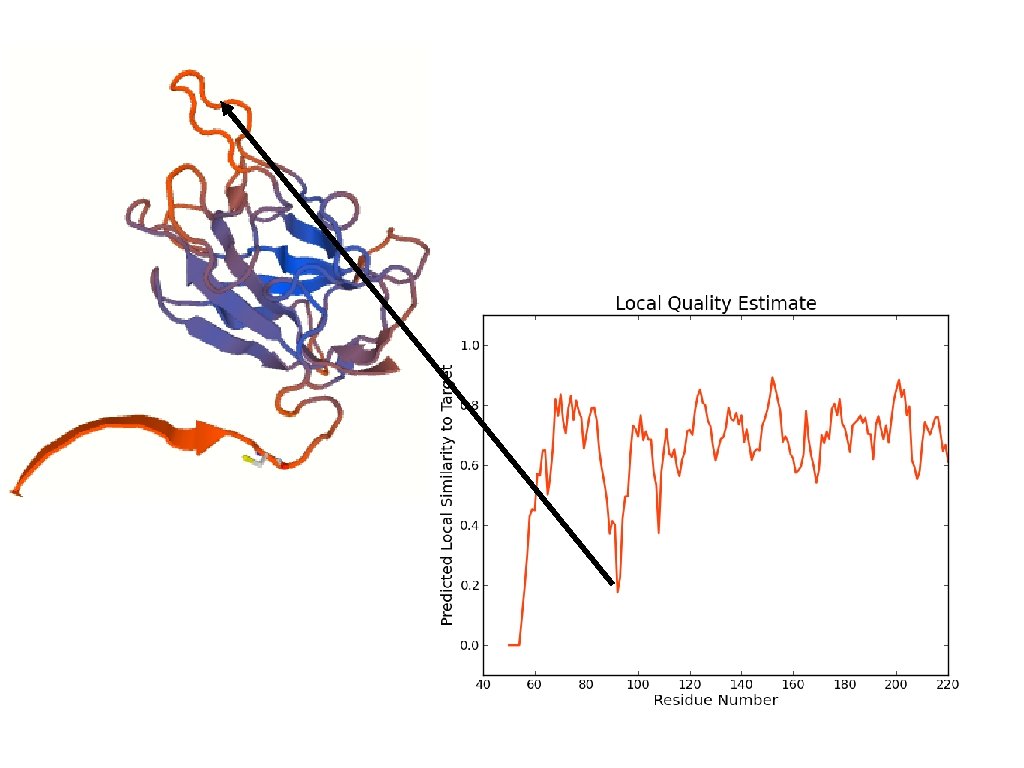
Assess your model(s). Download the PDB and submit it to Mol. Probity • Are you satisfied with the quality of this model here? If not, what strategies might further improve the modelling of this query?
Model evaluation - answers The quality of the model depends largely on the templat you choose. Moreover, differences in model quality can result from differences in sequence identity between templates (the higher the identity the better). To verify this try to use different templates and compare. Using X-ray structures as templates generally (not always) results in better models than using NMR or EM structures. Using better resolution templates (lower values) also normally results in better models.
• There is no “perfect” structure which satisfies every criteria • Try assessing an experimentally determined PDB structure!
Assessing Structure Models • Compare the structures generated from Automated Mode and Alignment Mode by looking at structure quality assessment from Swiss. Model and Mol. Probity • Superimpose the predicted structures generated from Automated Mode and Alignment Mode. Can you identify which is the better model? Useful pymol commands: • Super <p 1>, <p 2> • Align <p 1>, <p 2> for two objects p 1, p 2.
domain sequence Automated Mode SWISS MODEL extracted domain from entire sequence Uni. Prot find homologous sequences - BLAST template(s) selection multiple sequence alignment - t-coffee template(s) - target alignment Alignment Mode SWISS MODEL prediction Py. Mol tutorial Model assessment Mol. Probity Structural comparison Py. Mol assessment visual analysis
Useful links • http: //www. uniprot. org/ • https: //swissmodel. expasy. org/ • https: //blast. ncbi. nlm. nih. gov/Blast. cgi • https: //www. ebi. ac. uk/Tools/sss/ncbiblast/ • http: //tcoffee. crg. cat/ • https: //www. ebi. ac. uk/Tools/msa/tcoffee/ • http: //pfam. xfam. org/ • https: //www. ebi. ac. uk/interpro/