Protein metabolism II v Metabolism of Aromatic amino

Protein metabolism II v. Metabolism of Aromatic amino acids v. Metabolism of Sulphur containing amino acids v. Metabolism of Histidine v. Metabolism of Branched chain amino acids v. Metabolism of Arginine v. Aminoacidurias

Metabolism of aromatic amino acids

Aromatic amino acids v. Phenylalanine v. Tyrosine v. Tryptophan


Structure Phenylalanine CH 2 CH NH 2 COOH

Synthesis Essential amino acid - not synthesized in the body and has to be provided through the diet.

Structure Phenylalanine CH 2 CH COOH NH 2 Tyrosine HO CH 2 CH NH 2 COOH

Synthesis Tyrosine Non essential amino acid- synthesized in the body from phenylalanine

Synthesis O 2 Phenylalanine H 2 O Tyrosine Phenylalanine hydroxylase Tetrahydrobiopterin Dihydrobiopterin reductase NADP + NADPH+H +

Catabolism Tyrosine αKG Tyrosine transaminase PLP Glutamate Para hydroxy phenylpyruvate P- hydroxy phenyl pyruvate Vit C O 2 ++ hydroxylase (Cu ) CO 2 Dihydroxy phenylacetate (Homogentisic acid) Homogentisic acid oxidase (Fe 2+) Maleylacetoacetic acid Maleylacetoacetate isomerase Fumarylacetoacetic acid Fumarylacetoacetate hydrolase Fumarate TCA cycle Aceto acetate Ketogenic pathway Glucogenic pathway

Functions 1. Constituent of proteins: required for the protein synthesis

Functions 2. Synthesis of biologically important compounds §Catecholamines §Thyroid hormones §Melanin

Functions 2. Synthesis of biologically important compounds §Catecholamine's Epinephrine Norepinephrine Dopamine

Fright Flight Epinephrine Fight
 Synthesis of catecholamine's Site: In adrenal medulla & sympathetic ganglia Function: §")
Functions a) Synthesis of catecholamine's Site: In adrenal medulla & sympathetic ganglia Function: § Increase blood pressure §Increase contraction and rate of heart §Stimulates glycogenolysis and lipolysis §Dopamine is a inhibitor of prolactin secretion §Dopamine is a neurotransmitter in extrapyramidal tract, substantia nigra
 Synthesis of catecholamines Functions Tyrosine Tetrahydrobiopterin O 2 Tyrosine hydroxylase Dihydrobiopterin H 2")
a) Synthesis of catecholamines Functions Tyrosine Tetrahydrobiopterin O 2 Tyrosine hydroxylase Dihydrobiopterin H 2 O Dihydroxy phenylalanine (DOPA) PLP CO 2 DOPA decarboxylase Dopamine hydroxylase ++ Vit C, Cu Norepinephrine SAM N- methyl transferase SAH Epinephrine
Catabolism of catecholamines Tyrosine Dopamine Norepinephrine Epinephrine SAM Catechol-O- methyl transferase SAH Metanephrine")
Functions a)Catabolism of catecholamines Tyrosine Dopamine Norepinephrine Epinephrine SAM Catechol-O- methyl transferase SAH Metanephrine Mono amine oxidase Vanillyl mandelic acid (VMA) Excreted in urine (2 -6 mg/day)
Parkinsons disease: Neurological disease that results from insufficient formation and activity of")
Clinical significance: a)Parkinsons disease: Neurological disease that results from insufficient formation and activity of dopamine in brain b) Pheochromocytoma: is a rare tumor of adrenal gland (catecholamine secreting tumor derived from chromaffin cells) resulting in excess release of epinephrine.
 Synthesis of thyroid hormones Site: In thyroid gland under the influence of")
Functions b) Synthesis of thyroid hormones Site: In thyroid gland under the influence of TSH Functions §Increases basal metabolic rate (BMR) §Increases heart rate and force of contraction of heart muscle §Increases protein sythesis §Activates gluconeogenesis and glycogenolysis

Blood Thyroid gland Oxidation I T Iodide Thyro peroxidase + T Active iodine I _ 1 H 2 O 2 H 2 O

Thyroid gland I T 1 T + I _ 2 3 + I T + Protein 3 5 Protein + Iodination T Monoiodotyrosine + Diiodotyrosine (MIT) (DIT) I Blood

I T 1 T + 2 3 + I T + + Protein Iodination Monoiodotyrosine (MIT) 3 3 5 T Protein + I _ Thyroid gland I Blood Diiodotyrosine (DIT) Coupling Protein Tri iodo tyronine-Thyroglobulin 4 Proteolysis Tri iodo tyronine (T 3)

I T + 2 I + I T 3 3 5 1 + T Protein 5 + T Protein + I T T I _ Thyroid gland Diiodotyrosine +Diiodotyrosine Iodination (DIT) 3 Coupling I Blood Protein Tetra iodo tyronine-Thyroglobulin 4 Proteolysis Tetra iodo tyronine (T 4)

Melanin
 Synthesis of melanin Site: Deeper layers of skin (by melanocytes) under the")
Functions c) Synthesis of melanin Site: Deeper layers of skin (by melanocytes) under the influence of MSH
 Synthesis of melanin Function : gives black colour to skin, hair and")
Functions c) Synthesis of melanin Function : gives black colour to skin, hair and eyes. Protects the skin against harmful ultraviolet rays
 Synthesis of melanin Tyrosine ++ O 2 Tyrosinase (Cu ) H 2")
Functions c) Synthesis of melanin Tyrosine ++ O 2 Tyrosinase (Cu ) H 2 O Dihydroxy phenylalanine (DOPA) O 2 Tyrosinase H 2 O Dopaquinone Decarboxylation & oxidation Indolequinone Polymerisation Melanin
 Glucos Fat")
Summary Phenyl alanine Melanin Thyroid Hormones Tyrosine (T 3 & T 4) Glucos Fat Proteins e Catecholamine's (Epinephrine, Norepinephrine, Dopamine)
 2. Alkaptonuria 3.")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) 2. Alkaptonuria 3. Albinism 4. Tyrosinemia
 Autosomal recessive disorder")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Autosomal recessive disorder Incidence: 1 in 10, 000 births
 Biochemical defect Phenyalanine")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Biochemical defect Phenyalanine hydroxylase Phenylalanine Transamination Phenylpyruvate Reduction Decarboxylation Phenyl lactate Phenylacetate Conjugation (Glutamine) Phenylacetyl glutamine Tyrosine
 Clinical features CNS")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Clinical features CNS §Mental retardation §Seizures §Tremors §Hyperactivity §Delayed milestones
 Clinical features §Hypo")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Clinical features §Hypo pigmentation of skin and hair §Mousy body odour
 Diagnosis §Serum phenylalanine")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Diagnosis §Serum phenylalanine estimation >20 mg/dl (normal 2 -6 mg/dl) Detected by chromatography §Guthrie test Rapid screening test Bacterial bioassay for phenylalanine. Bacillus subtilis grows in culture medium containing phenylalanine §Ferric chloride test For detection of phenyl pyruvate in urine (Blue green colour +ve)
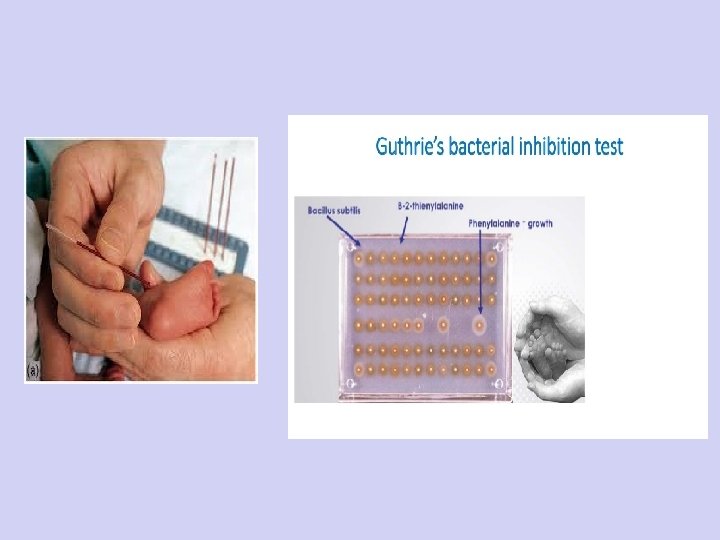
 Treatment §Early detection")
Inborn errors of phenylalanine and tyrosine metabolism 1. Phenyl ketonuria (PKU) Treatment §Early detection is important since mental retardation is preventable if detected at the earliest. (About 5 units of IQ lost for each 10 week delay in starting treatment) §Provide a special diet containing low phenylalanine (Tapioca based food has low phenylalanine) Special diet to be continued for the first decade of life , after which the child can have normal food

A 1 -year old girl presents with delayed mile stones, mousy odor , hypopigmentation. 1. Identify the inborn error of metabolism 2. What is the biochemical defect ?

Inborn errors of phenylalanine and tyrosine metabolism 2. Alkaptonuria Autosomal recessive disorder Incidence: 1 in 250, 000 births

Inborn errors of phenylalanine and tyrosine metabolism 2. Alkaptonuria Biochemical defect Homogentesic acid oxidase Homogentisic acid Maleyl aceto acetic acid Oxidation Urine & Polymerisation Alkaptone bodies (black pigment) Deposition in connective tissues (Bones, cartilage of nose, pinna of ear) (Ochronosis) Arthritis

Inborn errors of phenylalanine and tyrosine metabolism 2. Alkaptonuria Clinical features

Inborn errors of phenylalanine and tyrosine metabolism 2. Alkaptonuria Diagnosis Urine §Ferric chloride test +ve §Benedicts test+ve

Inborn errors of phenylalanine and tyrosine metabolism 2. Alkaptonuria Treatment Alkaptonuria is not a dangerous condition. So no specific treatment is required. However consumption of protein diet with relatively low phenylalanine content is recommended.

Inborn errors of phenylalanine and tyrosine metabolism 3. Albinism Autosomal recessive disorder Incidence: 1 in 20, 000 births

Inborn errors of phenylalanine and tyrosine metabolism 3. Albinism Biochemical defect Tyrosinase Tyrosine Melanin

Inborn errors of phenylalanine and tyrosine metabolism 3. Albinism Clinical features §Hypopigmentation of the skin, hair and eyes §Increased sensitivity to sunlight. Increased susceptibility to skin cancer §Photophobia (Intolerance to light)

Inborn errors of phenylalanine and tyrosine metabolism 3. Albinism Treatment Not specific Avoid exposure to UV rays

A 5 - year old girl has been brought by her mother for consultation. She has pale skin, blonde hair and pink iris. Parents reveal that their first two children have similar symptoms, but parents themselves are normal What is the probable diagnosis ?

Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia Autosomal recessive disorder Incidence: 1. 5 in 1, 000 births

Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia Types Type II
 Biochemical")
Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia type I (Hepato renal) Biochemical defect Deficiency of fumarylacetoacetate hydrolase
 Clinical")
Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia type I (Hepato renal) Clinical features §Cabbage like odour §Hypoglycaemia §Cirrhosis §Nephropathy
 Biochemical")
Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia type II (Oculo cutaneous) Biochemical defect Deficiency of Tyrosine transaminase
 Clinical")
Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia type. II (Oculo cutaneous) Clinical features §Mental retardation §Corneal lesions §Photophobia §Dermatitis

Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia Diagnosis §Increased serum tyrosine §Increased urinary excretion of tyrosine

Inborn errors of phenylalanine and tyrosine metabolism 4. Tyrosinemia Treatment Phenylalanine restricted diet

Phenylalanine Phenyl ketonuria Albinism Tyrosinemia type II P- hydroxy -phenylpyruvate Homogentisic acid Alkaptonuria Fumarylaceto acetic acid Tyrosinemia type I Fumarate Melanin


Structure CH 2 CH COOH Tryptophan N H NH 2

Synthesis Essential amino acid - not synthesized in the body and supplied through diet

Catabolism Tryptophan O 2 Tryptophan pyrrolase Kynurenine pathway N- formyl kynurenine THFA Kynurenine formylase One carbon Formyl THFA Kynurenine pool O 2 Kynurenine hydroxylase H 2 O 3 - hydroxy kynurenine Kynureninase Glucogenic Pyruvate Alanine (PLP) pathway 3 - hydroxy Anthranilic acid CO 2 NH 3 Aceto acetyl Co. A Acetyl Co. A Ketogenic pathway

Disorders related to tryptophan metabolism Xanthurenic aciduria

Disorders related to tryptophan metabolism 2. Xanthurenic aciduria Tryptophan Excreted in urine Xanthurenic acid 3 - hydroxy kynurenine Kynureninase (PLP) 3 - hydroxy Anthranilic acid

Putrefaction of Tryptophan Large intestine Skatole Indole SO 4 Indoxyl sulphate K + Indican Urine (Normal 2 -4 mg/day)

Functions 1. Constituent of proteins: required for the protein synthesis
 Serotonin b) Melatonin c) Niacin")
Functions 2. Synthesis of biologically important compounds a) Serotonin b) Melatonin c) Niacin
 Synthesis of serotonin Site: mast cells, platelets and gastrointestinal tract mucosa Functions:")
Functions a) Synthesis of serotonin Site: mast cells, platelets and gastrointestinal tract mucosa Functions: a)Brain- neurotransmitter, stimulates cerebral activity (excitation). Serotonin is involved in regulation of behavioral patterns, sleep, appetite and temperature b)Stimulator of smooth muscle contraction Vasoconstrictor GIT - increases motility of GIT Bronchoconstriction
 Synthesis of serotonin Tryptophan Tetrahydrobiopterin Dihydrobiopterin O 2 Tryptophan H 2 O")
Functions a) Synthesis of serotonin Tryptophan Tetrahydrobiopterin Dihydrobiopterin O 2 Tryptophan H 2 O hydroxylase 5 -hydroxytryptophan Decarboxylase CO 2 5 -hydroxytryptamine (Serotonin)
 Catabolism of serotonin Tryptophan Tetra hydro biopterin Di hydro biopterin O 2")
Functions a) Catabolism of serotonin Tryptophan Tetra hydro biopterin Di hydro biopterin O 2 Tryptophan H 2 O hydroxylase 5 -hydroxytryptophan Decarboxylase CO 2 5 -hydroxytryptamine (Serotonin) O 2 NH 3 Monoamine oxidase 5 -hydroxy indole acetate (5 -HIAA)
")
Disorders related to serotonin metabolism Malignant carcinoid syndrome (Argentaffinoma)

Disorders related to serotonin metabolism Malignant carcinoid syndrome Tumor of the argentaffin cells of the gastrointestinal tract , which synthesize serotonin Biochemical abnormality Increased amount argentaffin cells Increased serotonin synthesis (Increased diversion of tryptophan for serotonin synthesis)

Disorders related to serotonin metabolism Malignant carcinoid syndrome Clinical features §Sweating §Diarrhea §Hypertension §Pellagra like symptoms(since tryptophan is diverted for serotonin synthesis there is impaired synthesis of NAD) Diagnosis Increased excretion of 5 -hydroxy indole acetate(5 -HIAA) in urine
 Synthesis of melatonin Site: pineal gland in brain. Function: a) Melatonin involved")
Functions b) Synthesis of melatonin Site: pineal gland in brain. Function: a) Melatonin involved in diurnal variations of the body, sleep wake cycles and the biological rhythms. b) Melatonin blocks the production of melanocyte stimulating hormone(MSH) and adrenocorticotropic hormone (ACTH)
 Synthesis of melatonin Tryptophan Tetra hydro biopterin Di hydro biopterin O 2")
Functions b) Synthesis of melatonin Tryptophan Tetra hydro biopterin Di hydro biopterin O 2 Tryptophan H 2 O hydroxylase 5 -hydroxytryptophan Decarboxylase CO 2 5 -hydroxytryptamine (Serotonin) Acetyl Co. A Acetylase Acetyl serotonin SAM Methyl transferase SAH Melatonin
 Synthesis of niacin About 3% of tryptophan molecules are diverted at the")
Functions c) Synthesis of niacin About 3% of tryptophan molecules are diverted at the level of 3 - hydroxy anthranilate to form NAD. About 60 mg of tryptophan will be equivalent to 1 mg of niacin

Functions Synthesis of niacin Tryptophan N- formyl kynurenine Kynurenine 3 - hydroxy kynurenine 3 - hydroxy anthranilic acid Quinolinic acid CO 2 Nicotinic acid PRPP Quinolinate phospho ribosyl transferase (QPRT) Nicotinate mono nucleotide (NMN) ATP Nicotinamide adenine dinucleotide(NAD )
 Serotonin Melatonin 5 hydroxy indole")
Summary Diet Tryptophan Proteins Glucose Fat Coenzyme of niacin(NAD+) Serotonin Melatonin 5 hydroxy indole acetic acid

Inborn errors of tryptophan metabolism 1. Hartnup’s disease Autosomal recessive disorder

Inborn errors of tryptophan metabolism 1. Hartnup’s disease Biochemical defect Defect in transport of tryptophan and other neutral amino acids from the intestine, renal tubules and brain

Inborn errors of tryptophan metabolism 1. Hartnup’s disease Clinical features §Dermatitis-due to decreased niacin §Cerebral ataxia due to decreased serotonin §Mental retardation

Inborn errors of tryptophan metabolism 1. Hartnup’s disease Diagnosis Increased urinary excretion of indole compounds (Obermeyer test)

Inborn errors of tryptophan metabolism 1. Hartnup’s disease Treatment High protein diet Niacin supplementation

Metabolism of sulphur containing amino acids

sulphur containing amino acids Methionine Cysteine Cystine

 CH 2 S CH 3 CH 2 CH NH 2")
Structure Methionine Met (M) CH 2 S CH 3 CH 2 CH NH 2 COOH

Synthesis Essential amino acid - not synthesized in the body and has to be provided through the diet.
- activation of methionine 2. Conversion of methionine to")
Catabolism 1. Synthesis of S-adenosylmethionine (SAM)- activation of methionine 2. Conversion of methionine to cysteine 3. Regeneration of methionine
 CH 2 S CH 3")
Catabolism 1. Synthesis of S-adenosylmethionine (SAM / active methionine) CH 2 S CH 3 CH COOH NH 2 ATP Methionine CH 2 S CH 3 Adenosine CH Methionine COOH NH 2 SAM PPi +Pi Methionine H 2 O adenosyl transferase S-adenosylmethionine (SAM or active methionine)

Catabolism S-adenosylmethionine in transmethylation reaction Methionine ATP PPi +Pi Methionine H 2 O adenosyl transferase S-adenosylmethionine (SAM or active methionine) Acceptor Methylated Methyl transferase acceptor S-adenosyl homocysteine (SAH)

Ctabolism 2. Conversion of methionine to cysteine Methionine S-adenosylmethionine S-adenosyl homocysteine Adenosine homocysteinase adenosine Homocysteine Serine H 2 O Cystathionine β synthase Cystathionine H 2 O Cystathionase Cysteine + Homoserine (PLP)

Catabolism Degradation of cysteine Methionine S-adenosylmethionine S-adenosyl homocysteine Homocysteine Cystathionine Cysteine + Homoserine NH 3 α keto butyrate H 2 O CO 2 Propionyl Co. A Succinyl Co. A TCA cycle

Catabolism 2. Regeneration of methionine from S-adenosylmethionine ATP Methionine adenosyl transferase Folic acid Methyl THF Vit B 12 Methionine Homocysteine methyl transferase THF PPi +Pi SAM Acceptor Methyl transferase Methyl Vit B 12 Homocysteine Methylated SAH acceptor Adenosine homocysteinase Adenosine

Functions 1. Constituent of proteins Methionine is the first amino acid to be incorporated during protein biosynthesis, either as methionine (in eukaryotes) or formyl methionine (in prokaryotes)

Functions 2. S-adenosylmethionine: Principal donor of methyl group in transmethylation reactions in the body Active methyl group (Labile) CH 2 +S CH 3 Adenosine CH COOH NH 2 SAM S-adenosylmethionine (SAM or active methionine)
 Acceptor Methyl transferase Methylated acceptor S-adenosyl")
Functions Transmethylation reaction S-adenosylmethionine (SAM or active methionine) Acceptor Methyl transferase Methylated acceptor S-adenosyl homocysteine (SAH)
 Nor epinephrine Methyl transferase Epinephrine S-adenosyl homocysteine (SAH)")
Examples of transmethylation reaction S-adenosylmethionine (SAM) Nor epinephrine Methyl transferase Epinephrine S-adenosyl homocysteine (SAH)

Examples of transmethylation reactions Methyl Acceptor Methylated product Norepinephrine Epinephrine Guanido acetic acid Creatine Ethanolamine Acetyl serotonin Choline Melatonin

Significance of transmethylation reaction §Synthesis of biologically important compounds Egs: Epinephrine, creatine, choline, melatonin §SAM also serves as precursor for the synthesis of polyamines

Functions 3. Synthesis of cysteine Methionine S-adenosylmethionine S-adenosyl homocysteine Homocysteine Serine H 2 O Cystathionine β synthase Cystathionine H 2 O Cystathionase Cysteine + Homoserine (PLP)

Functions Methionine 4. Glucogenic role Homocysteine Cystathionine Cysteine + Homoserine Propionyl Co. A Succinyl Co. A TCA cycle Glucogenic role

Inborn Errors of Methionine Metabolism 1. Homocystinurias 2. Cystathioninuria

Inborn Errors of Methionine Metabolism 1. Homocystinurias Autosomal recessive disorder Incidence: 1 in 200, 000 births

Inborn Errors of Methionine Metabolism 1. Homocystinuria Types Homocystinuria type III Homocystinuria type IV

1. Homocystinuria Biochemical defect in type I Methionine Homocysteine Type I Cystathionine β synthase (PLP) Cystathionine

Inborn Errors of Methionine Metabolism 1. Homocystinuria Symptoms §Mental retardation §Skeletal deformities: due to interference of homocysteine with collagen cross linkages -Flat foot (Charlie chaplin gait) -Osteoporosis §Ectopia lentis (subluxation of lens) §Vascular thrombosis and coronary artery disease (Homocysteine activates hageman's factor and triggers platelet aggregation )

Inborn Errors of Methionine Metabolism 1. Homocystinurias

Inborn Errors of Methionine Metabolism 1. Homocystinurias Diagnostic tests §Plasma: Increased levels of homocysteine and methionine Detected by chromatography §Urine: Increased excretion of homocystine (Blood homocysteine levels are increased which combine to form homocystine which gets excreted) Cyanide nitroprusside test positive: due to increased excretion of homocystine

Inborn Errors of Methionine Metabolism 1. Homocystinuria Treatment §Restrict methionine and provide diet rich in cysteine (complete defeciency of cystathionine β synthase) §Provide pyridoxine in diet (activates cystathionine β synthase)

Inborn Errors of Methionine Metabolism 2. Cystathioninuria Autosomal recessive disorder Incidence: 1 in 200, 000 births
 Cysteine")
Inborn Errors of Methionine Metabolism 2. Cystathioninuria Biochemical defect Methionine Homocysteine Cystathionase (PLP) Cysteine

Inborn Errors of Methionine Metabolism 2. Cystathioninuria Symptoms §Mental retardation

Inborn Errors of Methionine Metabolism 2. Cystathioninuria Diagnostic tests §Plasma: Increased levels of cystathionine detected by chromatography §Urine: Increased excretion of cystathionine Cyanide nitroprusside test negative

Inborn Errors of Methionine. Metabolism 2. Cystathioninuria Treatment §Restrict methionine and provide diet rich in cysteine §Provide pyridoxine in diet (activates cystathionase)

Metabolism of cysteine
 CH 2 SH CH COOH NH 2")
Structure Cysteine Cys (C) CH 2 SH CH COOH NH 2

Synthesis Non essential amino acid - synthesized in the body Methionine S-adenosylmethionine S-adenosyl homocysteine Homocysteine Serine H 2 O Cystathionine β synthase (PLP) Cystathionine H 2 O Cystathionase (PLP) Cysteine + Homoserine

Catabolism 1. Transamination pathway 2. Oxidative pathway

Catabolism 1. Transamination pathway Cysteine Transaminase Mercapto pyruvate Sulfur transferase 2 H Pyruvate H 2 S
![Catabolism 2. Oxidative pathway Cysteine Dioxygenase [O 2] Cysteine sulfinate Transaminase Sulfinyl pyruvate Desulfinase](http://slidetodoc.com/presentation_image_h/2f4f58433a07865104f3da83a7521c10/image-119.jpg "Catabolism 2. Oxidative pathway Cysteine Dioxygenase [O 2] Cysteine sulfinate Transaminase Sulfinyl pyruvate Desulfinase")
Catabolism 2. Oxidative pathway Cysteine Dioxygenase [O 2] Cysteine sulfinate Transaminase Sulfinyl pyruvate Desulfinase 2 SO 3 Pyruvate
![Catabolism Metabolism of sulfur Cysteine H 2 S [O] Sulfide 2 SO 3 Sulfite](http://slidetodoc.com/presentation_image_h/2f4f58433a07865104f3da83a7521c10/image-120.jpg "Catabolism Metabolism of sulfur Cysteine H 2 S [O] Sulfide 2 SO 3 Sulfite")
Catabolism Metabolism of sulfur Cysteine H 2 S [O] Sulfide 2 SO 3 Sulfite [O] SO 4 2 - Sulfate Urine

Functions
 Protein structure Cysteine residues form the disulfide bonds, which")
Functions 1. Protein synthesis a) Protein structure Cysteine residues form the disulfide bonds, which is required for the structure of proteins Eg: §Primary structure of Insulin §Quaternary structure of immunoglobins b) Active sites of enzymes Cysteine is the active site of sulfhydryl enzymes Egs: Papain, cathepsin, glyceraldehyde 3 phosphate

Functions 2. Glucogenic role Cysteine Pyruvate Gluconeogenesis Glucose
![Functions 3. Synthesis of taurine Cysteine Dioxygenase [O 2] Cysteine sulfinate [O] Cysteic acid](http://slidetodoc.com/presentation_image_h/2f4f58433a07865104f3da83a7521c10/image-124.jpg "Functions 3. Synthesis of taurine Cysteine Dioxygenase [O 2] Cysteine sulfinate [O] Cysteic acid")
Functions 3. Synthesis of taurine Cysteine Dioxygenase [O 2] Cysteine sulfinate [O] Cysteic acid CO 2 Taurine

Functions of Taurine 1. Conjugation of bile acids Cholesterol Primary bile acids (Cholic acid, Chenodeoxycholic acid ) Conjugation with taurine Secondary bile acids (Taurocholic acid, Chenodeoxytaurocholic acid) 2. Acts as inhibitory neurotransmittor
 Mercapturic acid (Non")
Functions 4. Detoxification Cysteine + Acetic acid Acetyl cysteine Bromobenzene (Toxic) Mercapturic acid (Non toxic) Urine

Functions 5. Synthesis of glutathione 127
![Functions 6. Synthesis of phospho adenosine phospho sulfate(PAPS) 2 - [O] 2 H 2](http://slidetodoc.com/presentation_image_h/2f4f58433a07865104f3da83a7521c10/image-128.jpg "Functions 6. Synthesis of phospho adenosine phospho sulfate(PAPS) 2 - [O] 2 H 2")
Functions 6. Synthesis of phospho adenosine phospho sulfate(PAPS) 2 - [O] 2 H 2 S SO 3 Sulfide Sulfite SO 4 Sulfate 2 ATP 2 ADP+Pi PAPS Phospho adenosine phospho sulfate (Active sulfate) Functions of PAPS 1. Synthesis of glycosaminoglycans 2. Synthesis of sulfolipids 3. Detoxification of drugs

Functions 7. Synthesis of Coenzyme A Cysteine Decarboxylase CO 2 βmercaptoethanolamine Coenzyme A

Summary Proteins Diet Methionine SAM Glucose Glutathione Coenzyme A Proteins Cysteine PAPS Taurine Glucose Cystine Transmethylation

Inborn Errors of cystine Metabolism 1. Cystinuria 2. Cystinosis

Inborn Errors of cystine Metabolism 1. Cystinuria Autosomal recessive disorder Incidence: 1 in 7000 births

Inborn Errors of Cystine Metabolism 1. Cystinuria Biochemical defect Defective transporters in the kidney for amino acids cystine, lysine, arginine and ornithine, due to which kidneys fail to reabsorb them.

Inborn Errors of cystine Metabolism 1. Cystinuria Clinical features §Cystine calculi §Urinary obstruction and infection §Renal failure

Inborn Errors of cystine Metabolism 1. Cystinuria Diagnosis Plasma: Increased levels of cystine, detected by chromatography Urine: Increased excretion of cystine Positive cyanide nitroprusside test

Inborn Errors of cystine Metabolism 1. Cystinuria Treatment §Increase fluid intake §Sodium bicarbonate (alkalinizes urine and increases solubility of cystine)
 Autosomal recessive disorder")
Inborn Errors of cystine. Metabolism 2. Cystinosis (Cystine Storage Disease) Autosomal recessive disorder

Inborn Errors of cystine Metabolism 2. Cystinosis Biochemical defect Genetic defect in the lysosomal carrier mediated transport of cystine.

Inborn Errors of cystine Metabolism 2. Cystinosis Clinical features §Cystine calculi in various tissues §Renal failure

Inborn Errors of Cysteine Metabolism 2. Cystinosis Treatment §Increase fluid intake §Sodium bicarbonate (alkalinizes urine and increases solubility of cystine) §D-pencillamine (binds to cystine and excretes)


Structure CH 2 Histidine HN N CH NH 2 COOH

Synthesis Semi essential amino acid- not essential for normal adult, but needed to be supplied through the diet for children, pregnant and lactating women.

Catabolism Pyruvate Alanine Imidazole Transaminase pyruvic acid Histidine NH 3 Histidase Urocanic acid H 2 O Urine Urocanase Imidazole propionic acid Hydrolase H 2 O N-formimino glutamic acid (FIGLU) THFA Formimino transferase Glutamic acid Transaminase α keto glutarate Glucogenic pathway

Functions v. Protein synthesis Its pk value is 6. 8. Histidine is responsible for the buffering action of proteins.

Functions v. Synthesis of histamine Histidine CO 2 Decarboxylase Histamine Site: Mast cells, basophils and platelets Importance Actions of histamine are mediates through receptors (H 1, H 2)

Functions v. Synthesis of histamine Importance Actions of histamine are mediates through receptors (H 1, H 2) Actions of histamine through H 1 receptors Bronchial smooth muscle contraction, vasodilatation, allergic rhinitis Actions of histamine through H 2 receptors Gastric acid secretion

Functions Drugs affecting histamine metabolism: Antihistaminics Mechanism of action Antihistaminics Block histamine receptors Treatment of Allergic reactions (Citrezine) Treatment of Gastritis (Rantac)

Inborn errors of histidine metabolism 1. Histidinemia Autosomal recessive disorder Incidence: 1 in 20, 000 live births

Inborn errors of histidine metabolism 1. Histidinemia Biochemical defect Transaminase Increased Imidazole pyruvic acid Increased excretion in urine Histidase Urocanic acid

Inborn errors of histidine metabolism 1. Histidinemia Clinical features §Mental retardation §Delayed speech

Inborn errors of histidine metabolism 1. Histidinemia Diagnosis Elevated levels of plasma histidine Increased excretion of imidazole pyruvate and histidine in urine

Inborn errors of histidine metabolism 1. Histidinemia Treatment Diet restriction of histidine
 Folic acid THFA Formimino THFA Glutamic")
FIGLU excretion test Histidine N-formimino glutamic acid (FIGLU) Folic acid THFA Formimino THFA Glutamic acid α keto glutarate Glucogenic pathway
 Increased excretion in urine Folic acid")
FIGLU excretion test Histidine N-formimino glutamic acid (FIGLU) Increased excretion in urine Folic acid THFA Glutamic acid Indicator of folic acid deficiency


Structure CH 2 NH Arginine C CH 2 NH NH 2 CH 2 NH C NH 2 CH COOH NH 2 NH CH 2 CH NH 2 COOH

Synthesis Semi essential amino acid- not essential for normal adult, but needed to be supplied through the diet for children, pregnant and lactating women.

Catabolism Arginine urea Ornithine Arginase Transaminase Glutamate semialdehyde Dehydrogenase Glutamate Dehydrogenase α keto glutarate Glucogenic pathway
")
Functions v. Synthesis of biologically important compounds Creatine Polyamines Nitric oxide (NO)

Functions v. Synthesis of polyamines
 Stimulate protein and nucleic acid biosynthesis b) Required")
Functions v. Importance of polyamines a) Stimulate protein and nucleic acid biosynthesis b) Required for cell proliferation c) Stabilization of ribosome's and DNA
 Chemistry: Highly reactive free radical Site: Neurons,")
Functions v. Synthesis of Nitric oxide (NO) Chemistry: Highly reactive free radical Site: Neurons, macrophages and endothelium
 Arginine O 2 H 2 O NADPH+H")
Functions v. Synthesis of Nitric oxide (NO) Arginine O 2 H 2 O NADPH+H NADP + Nitric oxide + Citrulline + Nitric oxide synthase
 Blood vessels-Vasodilator (decreases blood pressure) b) Smooth")
Functions v. Function of nitric oxide a) Blood vessels-Vasodilator (decreases blood pressure) b) Smooth muscle- relaxation c) CNS –neurotransmittor d) Platelets-inhibit platelet aggregation

Functions v. Clinical importance Angina pectoris Nitroprusside Synthesis nitric oxide Dilate coronary arteries

Functions v. Clinical importance Pulmonary hypertension Nitric oxide inhalation Dilate pulmonary arteries Reduce pulmonary oedema
 Nitric oxide synthesis Relaxes the smooth muscle")
Functions v. Clinical importance Impotence Sildenafil (Viagra) Nitric oxide synthesis Relaxes the smooth muscle in corpus cavernosum Increases blood flow to penis Treat erectile dysfunction


Structure H 3 C Valine Leucine CH H 3 C COOH NH 2 H 3 C CH H 3 C Isoleucine CH CH 2 CH COOH NH 2 H 3 C H 2 C CH H 3 C CH NH 2 COOH

Synthesis Essential amino acid-not synthesized in the body and has to be provided through the diet.

Catabolism Branch chain amino acids Transaminase Corresponding branch chain α keto acids α keto acid dehydrogenase Corresponding branch chain Fatty acyl Co. A Succinyl Co. A Glucogenic pathway (Valine) Acetoacetyl Co. A Acetyl Co. A and propionyl Co. A Ketogenic Glucogenic and ketogenic pathway pathway (Leucine) (Isoleucine)
 Autosomal")
Inborn errors of branch chain amino acids metabolism Maple syrup urine disease (MSUD) Autosomal recessive disorder Incidence: 1 per 1 lakh live births
 Biochemical")
Inborn errors of branch chain amino acids metabolism Maple syrup urine disease (MSUD) Biochemical defect Branch chain amino acids Increased excretion in urine Corresponding branch chain α keto acids Branched chain α keto acid dehydrogenase Corresponding branch chain fatty acyl Co. A
 Clinical")
Inborn errors of branch chain amino acids metabolism Maple syrup urine disease (MSUD) Clinical features Symptoms appear within the first week of life and death may occur in the first year of life if not treated §Vomiting §Mental retardation §Convulsions §Acidosis §Coma §Urine: Maple syrup smell (Burnt sugar smell)
 Diagnosis v. Increased urinary")
Inborn errors of tryptophan metabolism Maple syrup urine disease (MSUD) Diagnosis v. Increased urinary excretion of branch chain α keto acids
 Treatment Giving diet low")
Inborn errors of tryptophan metabolism Maple syrup urine disease (MSUD) Treatment Giving diet low in branch chain amino acids

Case study A neonate presented at day 12 with metabolic acidosis, abnormal urine odor, ketonuria and hepatosplenomegaly. Blood and urine studies revealed elevated levels of valine, isoleucine and leucine

Aminoacidurias Biochemical defect §An inherited defect in certain enzymes of amino acid metabolism §Defect in the absorption and transport of amino acids
 §Diagnosed by chromatography")
Aminoacidurias Diagnosis estimation of levels of amino acid (Elevated than normal) §Diagnosed by chromatography §Diagnosed by specific tests Urine Increased excretion of amino acid or its metabolite §Diagnosed by specific tests

Disorder of phenylalanine and tyrosine metabolism Disorder Biochemical Clinical Defect features Diagnosis Treatment 1. Phenyl Phenyalanine MR, ketonuria hydroxylase Seizures Tremors, Hypopigm entation Mousy odour of sweat Serum: phenylalanine Urine: phenyl pyruvate Diet restriction of phenylalan ine 2. Homogentesic The urine Serum: Alkapton acid oxidase becomes Homogentesic uria black on acid standing Urine: Arthritis Homogentesic acid Diet restriction of phenylalan ine & tyrosine

Disorder of phenylalanine and tyrosine metabolism Disorder Biochemical Clinical Defect features 3. Albinism tyrosinase Diagnosis Treatment Hypo Serum: Diet pigmentati tyrosine restriction on Urine: tyrosine of Photopho phenylalan bia ine & tyrosine 4. fumarylaceto MR, Serum: Tyrosinem acetate hepatoren tyrosine, ia I hydrolase al damage methionine Urine: tyrosine, PHPPA Diet restriction of phenylalan ine & tyrosine

Disorder of phenylalanine and tyrosine metabolism Disorder Biochemical Defect 5 Tyrosinem transaminase ia II Clinical features MR, corneal lesions Photopho bia Diagnosis Treatment Serum: Diet tyrosine restriction Urine: tyrosine of phenylalan ine & tyrosine

Sulphur containing amino acids metabolism disorder Disorde Biochemical r Defect Clinical features Diagnosis Treatment 6. Cystathionine MR, Homocy β ectopia stinuria synthase lentis, osteoporos is, vascular thrombosis Serum: Diet Homocystine, restriction methionine of Urine: methionine Homocystine 7. Cystathionase MR, Cystathi anemia oninuria Serum: cystathionine, Urine: cystathionine 8. Defective Cystinuri transporters in calculi a the kidney Renal failure Serum: cystine, Urine: cystine Diet restriction of methionine

Sulphur containing amino acids metabolism disorder Disorder Biochemical Defect 9. lysosomal Cystinosis carrier mediated transport of cystine. Clinical features Cystine calculi Renal failure Diagnosis Treatment Serum: cystine, Urine: cystine

Disorder of branch chain amino acids metabolism Disorder Biochemical Defect 10. Maple Branched syrup chain α keto urine acid dehydro disease genase Clinical features MR, Convulsion s Acidosis Coma Maple syrup smell of urine Diagnosis Treatment Serum: val, leu , isoleu, α keto acids Urine: val, Leu , isoleu, α keto acids Diet restriction of val, leu , isoleu

Disorder of histidine metabolism Disorder Biochemical Defect 11. Histidase Histidine mia Clinical features MR, speech defect Diagnosis Treatment Serum: histidine Urine: imidazole pyruvic acid Diet restriction of histidine

Disorder of tryptophan metabolism Disorder Biochemical Clinical Defect features 12. Defect in MR, Hartnups absorption Dermatitis disease and transport of tryptophan Diagnosis Treatment Serum: tryptophan Urine: indole compounds

Disorder of proline metabolism Disorder Biochemical Defect 13. Proline Hyperproli oxidase nemia Clinical features MR, renal damage Diagnosis Treatment Serum: Proline Urine: hydroxy proline Diet restriction of proline

Glycine metabolism disorder Disorder Biochemical Defect 14. defect in the Glycinuria renal tubular reabsorption Clinical features oxalate renal stones 15. Glycine oxalate Primary transaminase renal Hyperoxal stones uria Diagnosis Urine: glycine Serum: oxalate Urine: oxalate Treatment

Urea cycle disorder Disorder Biochemical Clinical Defect features Diagnosis Treatment 16 Carbamoyl Hyperam phosphate monemia synthase I Type 1 MR Intermittent ataxia Irritability Lethargy Vomitting Serum urine: Diet Ammonia, restriction glutamine of proteins urine: glutamine 17 Ornithine Hyperam transcarbam monemia oylase Type 2 MR Intermittent ataxia Irritability Lethargy Vomitting Serum urine: Diet Ammonia, restriction glutamine of proteins urine: glutamine

Urea cycle disorder Disorder Biochemical Defect 18. arginosuccin Citrullinem ate synthase ia Clinical features Irritability Lethargy Vomitting Diagnosis Treatment Serum: Ammonia, citrulline Urine: citrulline Diet restriction of proteins 19. arginosuccin Argininosu ase ccinicacid uria Irritability Lethargy Vomitting Serum: Diet arginosuccin restriction ate , citrulline of proteins Urine: arginosuccin ate , citrulline

Urea cycle disorder Disorder Biochemical Clinical Defect features 20. Arginase Hyperargi ninemia Irritability Lethargy Vomitting Diagnosis Treatment Serum: arginine, Urine: arginine Diet restriction of proteins

- Slides: 194