Protein Analysis Hosted By Darren G Thifault The

Protein Analysis Hosted By: Darren G. Thifault
 Format • Standard representation for macromolecular structure data. •")
The Protein Data Bank (PDB) Format • Standard representation for macromolecular structure data. • The data can include; • • • Atomic Coordinates Crystallographic Structure Factors NMR Experimental Data Names of Molecules Primary and Secondary Structure Information Sequence Database References Ligand Biological Assembly Information Details about Data Collection and Structure Solution Bibliographic Citations

Protein Modelling Methods De Novo • Builds models from physicochemical parameters and neural net algorithms. • Limited to small proteins. • Require vast computational resources. • • • Threading Builds models from amino acid sequence and structural information of homologous protein templates. Used for targets with only fold-level homology. Can make predictions based on the structure information of sequences when no significant homology is found. Requires medial computational resources.

694 187 886 192 1582 696 2871 1289 3812 941 4985 1173 6550 1565 8608 2058 10964 2356 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2823 2999 4152 5151 5346 6413 7141 6909 7295 7761 7950 8775 9364 9598 9301 9861 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 2013 2014 2015 2016 124430 114569 105268 95670 86306 77531 69581 61820 54525 47616 40475 34062 Yearly 28716 23565 19413 16414 2627 13591 507 142 RCSB Protein Data Bank Statistics YEARLY GROWTH OF TOTAL STRUCTURES Total

CASP Results
")
I-TASSER (Iterative Threading ASSEmbly Refinement)
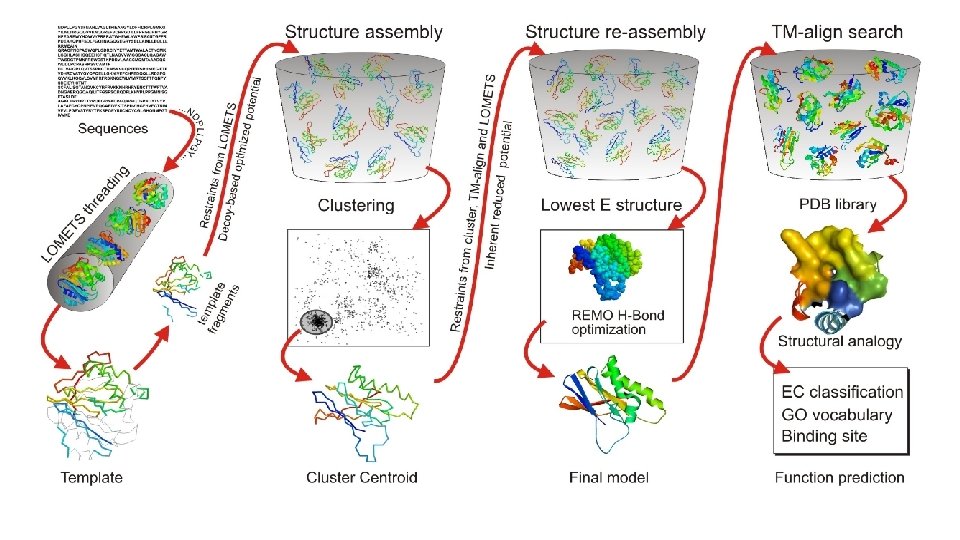

LOMETS Threading • Query sequence is aligned to known protein sequences in the NCBI Database using PSI-Blast to search and identify structural templates. • LOMETS consists of 8 threading algorithms (PPAS, Env-PPAS, w. PPAS, d. PPAS 2, wd. PPAS, MUSTER and w. MUSTER) which search for various structural features that account for their scoring matrix.

Template Fragments • Fragments of the aligned sequence templates are ranked to evaluate the significance of the alignment compared to the other templates. • >8 Templates suggests good homology • 1 -8 Templates suggests medium homology • 0 Templates suggests no homology
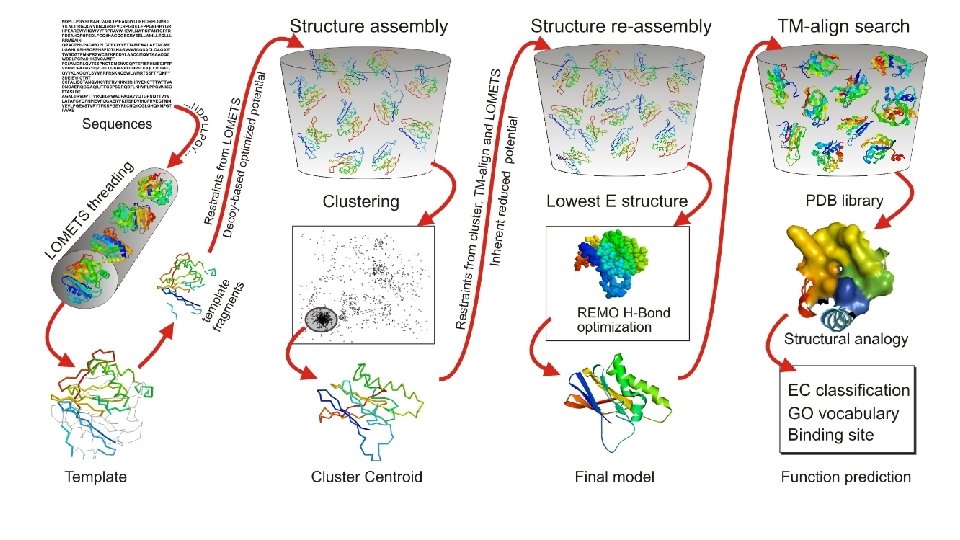

Structure Assembly • Query sequence is split into aligned and unaligned regions. • Aligned regions with >5 residues of a template fragment are kept frozen while the unaligned regions are built with ab initio simulations and serve as linkage for the threaded fragments. • Structure folding and reassembly are then performed using replica-exchange Monte Carlo simulations with optimized knowledge-based force fields consisting of three major components: (i) generic statistical potentials, (ii) hydrogen-bonding networks and (iii) threading-based restraints from LOMETS. • Output is a series of Decoy models.

Clustering • Decoy models from the structure assembly simulations are then clustered by their free -energy state for analysis by SPICKER. • A Centroid Model from each major cluster is generated by averaging the coordinates of all the conformations.
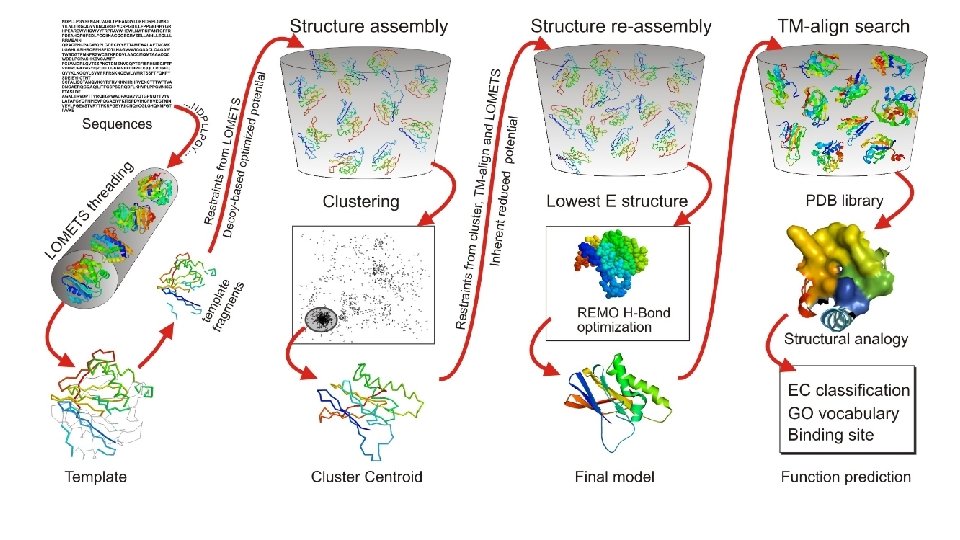

Structure Re-Assembly • Centroid cluster models are used in a second structure assembly simulation to remove any steric clashes and further refine the global topology. • Spatial restraints are added from structure templates detected by searching the PDB library with TM-align for structures that are similar to the cluster centroids from the first round of simulations.

Optimization • Final atomic model is constructed by Mod. Refiner starting from the low-energy conformations selected from the second round simulation trajectories. • In Mod. Refiner, the backbone structure is first built from the Cα-traces. Side-chain atoms are then constructed from a rotamer library with the full-atomic conformation refined by energy minimizations based on a composite physics and knowledge-based force field. • Global quality estimations are given through calculations of a confidence score (C-score). • Residue-level local quality estimations are calculated by Res. Q on the variation of modeling simulations and the uncertainty of homologous alignments.
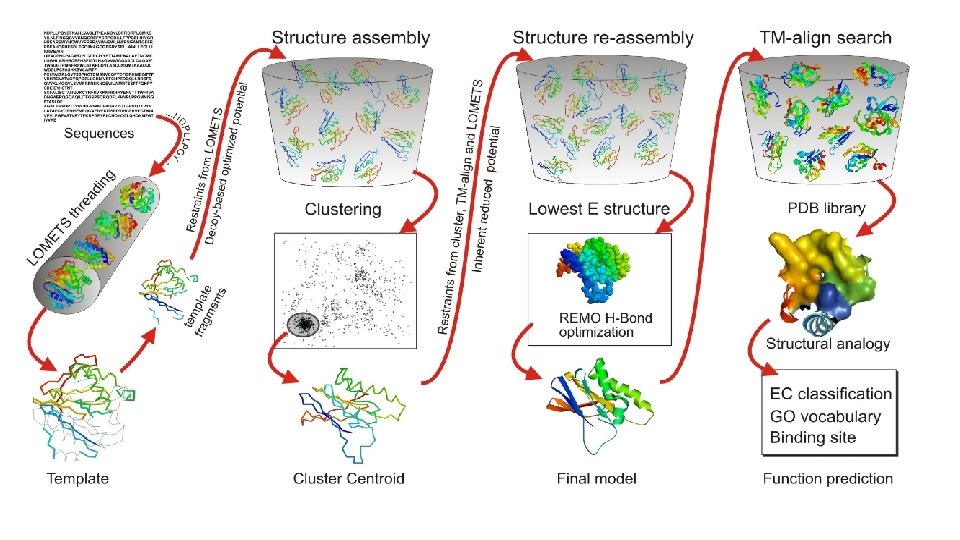

Function Prediction • Models with the highest confidence scores are matched against the Bio. Li. P 5 database of ligand-protein interactions to detect homologous function templates. • Functional insights on ligand-binding site (LBS), Enzyme Commission (EC) and Gene Ontology (GO) are predicted from the functional templates. • Three complementary algorithms (COFACTOR, TM-SITE and S-SITE) are used to enhance function inferences, the consensus of which is derived by COACH 4 using support vector machines.

Protein-Protein Docking
 • Internal Geometry of Protein is Fixed. •")
Docking Methods Rigid Body (Clus. Pro) • Internal Geometry of Protein is Fixed. • Require medial computational resources. • No Experiment Data Required. Flexible Body (HADDOCK) • Internal Geometry of Protein is Not Fixed. • Bond Angles • Bond Lengths • Torsion Angels • Require vast computational resources. • Experimental Data Required (NMR, Mutagenesis, etc. )

Questions?
- Slides: 20