PROTECTING HUMAN SUBJECTS IN RESEARCH WHY IS THIS

PROTECTING HUMAN SUBJECTS IN RESEARCH: WHY IS THIS IMPORTANT? Joey Casanova, BBA, CIP Associate Director for Educational Initiatives and Community Outreach Human Subject Research Office University of Miami

WHAT IS AN INSTITUTIONAL REVIEW BOARD? A group of people who review and approve research proposals involving human subjects Purpose: To protect the rights and welfare of human subjects
 & Non-Scientists")
WHO ARE THE MEMBERS OF AN IRB? • Scientists (MD, Ph. D) & Non-Scientists • Prisoner representative • Child advocate • Community member

WHAT DOES AN IRB DO? • Approve, Disapprove, or Modify • Conduct Continuing Review • Observe/Monitor/Audit • Suspend or Terminate Approval

SECTION 2 WHAT IS HUMAN SUBJECT RESEARCH?

DEFINITIONS OF “HUMAN SUBJECT” • HHS: A living individual about whom an investigator… conducting research obtains 1. Data through intervention or interaction with the individual, or 2. Identifiable private information • FDA: Recipient of test article or control

DEFINITIONS OF RESEARCH • HHS: A systematic investigation designed to develop or contribute to generalizeable knowledge • FDA: Clinical Investigations of FDA Regulated Products – any experiment in which a drug, biologic or significant risk device is administered or dispensed to or used involving one or more human subjects

IS IT “RESEARCH WITH HUMAN SUBJECTS”? Human Research Subjects

SECTION 3 HISTORY OF HUMAN SUBJECT PROTECTIONS
 1. 2. 3. 4. 5. 6. Nazi physicians on trial for")
NUREMBERG CODE (1947) 1. 2. 3. 4. 5. 6. Nazi physicians on trial for research atrocities 7. performed on prisoners of war. This resulted in 8. the first internationally recognized code of research ethics, issued by the Nazi War Crimes 9. Tribunal. 10. Voluntary Consent Fruitful results otherwise unobtainable Animal experiments Avoid physical & mental suffering Not done if injury expected Risk less than importance of the problem Protect subject from injury Qualified personnel Termination by subject Termination by investigator
 Kefauver-Harris Amendments: • Required preclinical testing using animals • Required IND")
THALIDOMIDE EXPERIENCE (1962) Kefauver-Harris Amendments: • Required preclinical testing using animals • Required IND submission to FDA for human studies • Ethical Scientific pre-review – not required • Informed Consent of human subjects – loosely required 28 FR 179 (Jan. 8, 1963)
 REVISIONS BASIC PRINCIPLES • Research based on animal experiments •")
DECLARATION OF HELSINKI (1964) REVISIONS BASIC PRINCIPLES • Research based on animal experiments • Conducted only by qualified persons • Importance of research proportionate to risk • Risks-benefits assessed before hand • Effects of drugs on personality considered • 1975 – Tokyo – Independent oversight – Informed consent • 1983 – Venice – “Consent” from minors • 1989 – Hong Kong – Function and structure of committee • 1996 – South Africa – Use of a placebo • 2000 – Edinburgh – Cognitively impaired – Conflicts of Interest – Best proven therapy
 • Public Health Service funded study to understand the")
TUSKEGEE SYPHILIS STUDY (1932– 72) • Public Health Service funded study to understand the natural progression of untreated syphilis in human beings. • The research population included approximately 300 indigent African-American sharecroppers in Macon County, Alabama. • The subjects thought they were receiving beneficial medical care. • The subjects were followed, untreated, many years after penicillin was known to cure syphilis.

CONGRESSIONAL HEARINGS ON THE QUALITY OF HEALTH CARE AND HUMAN EXPERIMENTATION Wichita Jury Study (50’s) Willowbrook Hepatitis Study (50’s) Jewish Chronic Disease Studies (60’s) Milgram Studies of Obedience to Authority (60’s) San Antonio Contraceptive Study (70’s) Tearoom Trade Study (70’s)
 • The National Commission for the Protection of Human Subjects")
NATIONAL RESEARCH ACT (1974) • The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research • “Conduct a comprehensive investigation… to identify the basic ethical principles which should underlie the conduct of… research involving human subjects…” Public Law 93 -348, Sec. 202(a)(1)(a)
 • RESPECT FOR PERSONS – Acknowledge and protect autonomy – Prevent")
BELMONT REPORT (1979) • RESPECT FOR PERSONS – Acknowledge and protect autonomy – Prevent coercion or undue influence • BENEFICENCE – Do unto others as you would have them do unto you • JUSTICE – Equal distribution of risks and potential benefits among those who may benefit

“NEW REGULATIONS” FOR PROTECTION OF HUMAN SUBJECTS HHS 45 CFR 46, Subpart A, B, C, D FR 45 (Jan. 26, 1981), p 8386 FDA 21 CFR 50 (Informed Consent) FDA 21 CFR 56 (Institutional Review Board) FR 46 (Jan. 27, 1981), p 8942 Revised (1991) to correspond to DHHS regulations

OTHER REGULATIONS, POLICIES AND GUIDELINES THE IRB MUST FOLLOW UM Policies/ Procedures State and Local Laws Federal Regulations • Veteran’s Affairs 38 CFR 16 • Federal Agency Policy • ICH (Good Clinical Practice) • Joint Commission on Accreditation of Healthcare Organizations (JCAHO) • Family Educational Rights and Privacy Act (FERPA) • International policies and ethic codes • Other funding agencies

HUMAN SUBJECTS PROTECTION: A SHARED RESPONSIBILITY IRB Investigators • Chair • Members • Staff • PI • Co-investigators • Staff Institution • Institutional Officials • Leadership Sponsors Regulators

SECTION 4 IRB REVIEWS

REVIEW CONTINUUM Exempt Expedited Full Low Minimal Higher Risk *Level of risk helps determine route of review
 & FDA 21 CFR")
WHAT IS MINIMAL RISK? • DHHS 45 CFR 46. 102(i) & FDA 21 CFR 56: – The probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during routine physical or psychological exams or tests • OHRP Guidance: – Minimal risk is relative to the daily life of a normal, healthy person. The risk threshold cannot increase because the person is sick and faces greater risk due to illness

“IT IS NOT INVASIVE SO IT MUST BE MINIMAL RISK” This is not correct! Risk of Criminal/Civil Liability Financial Risk Employment Risk of Stigmatization Risk to Insurability Risk of Embarrassment

EXEMPT RESEARCH • Six categories defined by 45 CFR 46 • Research must fall into one or more of the categories to be exempt • IRB has the responsibility to determine exemption, PI cannot make determination • May still require consent or other safeguards • How is data being collected? – Is there a code that links data to subjects (easily identifiable)?
 Educational research (2 -3) Tests, surveys, interviews or public observation (4)")
EXEMPTION CATEGORIES (1) Educational research (2 -3) Tests, surveys, interviews or public observation (4) Research on existing public or anonymous data or specimens (5) Federal demonstration projects (6) Taste and food evaluation

EXPEDITED REVIEW • Expedited review procedure may be used to review either : – Some or all of the research appearing on the list and found by the reviewer(s) to involve no more than minimal risk – Minor changes in previously approved research during the period (of one year or less) for which approval is authorized • Expedited does not mean quicker – rigor of review is the same, number of reviewers different • Review carried out by IRB chair or designee • Reviewers may approve or modify, not disapprove
 (1) Clinical studies: IND/IDE not required (2) Blood")
ELIGIBLE FOR EXPEDITED REVIEW: (INITIAL REVIEW) (1) Clinical studies: IND/IDE not required (2) Blood sample collection (3) Prospective collection of biological samples (noninvasive means) (4) Data collected through noninvasive means (routinely practiced in clinical settings) (5) Materials (data, documents, specimens) have been collected or will be collected for non-research purposes (6) Collection of voice, video or digital data (7) Individual or group behavior, surveys, interviews, oral histories
 • (8) Continuing review of research with no")
ELIGIBLE FOR EXPEDITED REVIEW: (CONTINUING REVIEW) • (8) Continuing review of research with no further direct subject participation – Long-term follow up for survival – No subjects have been enrolled – Data analysis • (9) Continuing review of minimal risk research (not under IND or IDE) where no additional risks have been identified

FULL REVIEW MEANS: • A full quorum is assembled (at least half of the members plus one, includes nonscientist) • All members participate in discussion and make comments (plenary review) • Decision is rendered by a majority of the assembled quorum • No member with a conflict of interest participates in the decision • Numerical vote is taken and recorded
 Reasonable risk/benefit assessment Equitable subject")
CRITERIA FOR APPROVAL • • Minimized risks (not eliminated) Reasonable risk/benefit assessment Equitable subject selection Informed consent process Informed consent documentation Data monitored for safety Confidentiality/privacy maintained Vulnerable populations protected

IRB APPROVAL CRITERIA & THE BELMONT REPORT Beneficence Justice Risk/Benefit Analyses Data Safety Subject Selection Inclusion/Exclusion Recruitment Methods Design of Experiment Qualifications of PI/Staff Respect for Persons Informed Consent Privacy & Confidentiality Vulnerable Populations Surrogate Consent Assent

SECTION 5 VULNERABLE POPULATIONS

VULNERABLE POPULATIONS • 45 CFR 46 includes specific protections: – Subpart B – Additional protections for Pregnant Women, Human Fetuses and Neonates Involved in Research (revised Dec. 13, 2001) – Subpart C – Additional DHHS Protections Pertaining to Biomedical and Behavioral Research Involving Prisoners as Subjects – Subpart D – Additional DHHS Protections for Children Involved as Subjects in Research • In addition, the regulations [§ 111(b)] require additional protections for any other subjects “…likely to be vulnerable to coercion or undue influence”

• • OTHER VULNERABLE POPULATIONS Decisionally or cognitively impaired Non-English speaking Desperately ill Economically disadvantaged Educationally disadvantaged Students Employees

SECTION 6 INFORMED CONSENT

CONSENT MUST BE: • Individual • Voluntary • Informed • Effective • Continuous

REQUIRED ELEMENTS • Study involves research – Purpose of the research – Duration of participation – Procedures to be followed – Procedures which are experimental • Foreseeable risks and discomforts • Reasonably expected benefits o 45 CFR 46. 116, 21 CFR 50. 25
 Alternative procedures Protection of confidentiality For research involving greater than")
REQUIRED ELEMENTS (CONT. ) Alternative procedures Protection of confidentiality For research involving greater than minimal risk: compensation/treatment, if any, for research-related injury Contacts for questions about the research, research-related injury, subjects’ rights Voluntary participation, refusal without loss of benefits, withdraw at any time o 45 CFR 46. 116, 21 CFR 50. 25
 • Unforeseeable risks to subject (and fetus) • Anticipated reasons")
ADDITIONAL ELEMENTS (WHEN APPROPRIATE) • Unforeseeable risks to subject (and fetus) • Anticipated reasons for termination by investigator • Additional costs to subjects • Consequences of withdrawal by participant • New findings • Number of subjects o 45 CFR 46. 116, 21 CFR 50. 25

SUBJECT RECRUITMENT • Recruitment ads, telephone scripts – Often the first step in informed consent – Must be reviewed by the IRB

CONSENT: PROCESS AND FORM Consent is a PROCESS in which ◦ ◦ Investigator discloses all relevant information Potential subject has opportunity to ask questions Investigator answers questions Repeat the above… The consent FORM is a permanent record of ◦ Information conveyed ◦ Subject’s willingness to participate

CONSENT PROCESS MUST Involve dynamic and continuing exchange of information between the study team and the participant Provide sufficient information to make informed choices about whether to begin or continue participation in clinical research Give subject enough time to consider the decision* Minimize possibility of coercion or undue influence* o* 45 CFR 46. 116, 21 CFR 50. 20

WHEN? Both FDA & ICH require that consent be obtained “prior to” a prospective subject’s participation in a clinical study. ◦ D/C existing medication for washout ◦ Performing study-specific screening exams or procedures ◦ Administering any test products Should verify documentation, understanding, and willingness to continue at every visit

ADEQUATE TIME? Use “reasonable man” criterion GCP: A Question & Answer Reference Guide, 2003 ◦ Time for person 6 th grade education to read form ◦ Time to question staff and consult friends or family ◦ Time, if requested, to review alternatives ◦ Time to deliberate and make decision “Time-out” – give pager # and allow privacy Send ICF in advance if possible Avoid requiring mass or immediate enrollment if possible

SPECIAL CONSIDERATIONS Emergency or Compassionate Use Biological Sample collection/banking Decisionally challenged subjects Exceptions HIV testing Genetic research Vulnerable populations

CONSENT FORM MUST… • Be understandable to subject or representative • NOT – Waive subject’s rights – Release investigator, sponsor, institution from liability o 45 CFR 46. 116, 21 CFR 50. 20

CRITERIA FOR WAIVING OR ALTERING INFORMED CONSENT • No more than minimal risk to subjects; • Waiver would not adversely affect the rights and welfare of subjects; • Could not be “practicably” carried out without the waiver or alteration; AND • When appropriate, subjects will be given additional pertinent information after participation. o 45 CFR 46. 116(d), but not FDA

DOCUMENTATION Use written form approved by IRB ◦ Embodies required elements ◦ Signed and dated by subject or legally authorized representative ◦ Give copy to subject/representative Short form ◦ States the required elements have been presented orally ◦ Witness needed ◦ IRB approves summary signed by witness ◦ Copy of short form and summary to subject o 45 CFR 46. 117, 21 CFR 50. 27

CRITERIA FOR WAIVER OF REQUIRED DOCUMENTATION 1. The only record linking the subject to research would be the consent document, and the principal risk relates to breach of confidentiality; OR 2. No more than minimal risk and involves no procedures for which consent is required outside of the research context.

WRITING THE CONSENT DOCUMENT • Written in 2 nd person. • Not valid unless the consenter understands the information that has been provided. • Avoid technical terms – Simple sentences • Translation is required when English is not participants’ primary language
![REMINDERS • Remove template instructions, for example [INSERT if applicable]. • Complete all sections](http://slidetodoc.com/presentation_image_h/c390768dd60b895725764d9553d421f4/image-51.jpg "REMINDERS • Remove template instructions, for example [INSERT if applicable]. • Complete all sections")
REMINDERS • Remove template instructions, for example [INSERT if applicable]. • Complete all sections unless not applicable. • If you are using a modification of the subject injury language, submit a copy of the contract so that the IRB can verify language.

SUGGESTIONS FOR ASSENT DOCUMENTS • Federal regulations allow for child to sign form in same language as parent, however, IRB recommends simplifying as much as possible. • Would obtain signature from child. • Discuss confidentiality issues, will parents know outcomes?

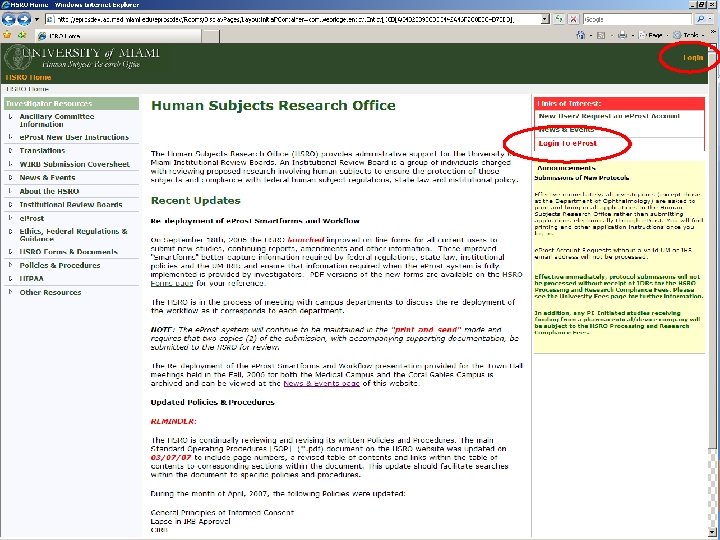
http: //hsro. miami. edu SECTION 7 EPROST REDESIGNED

EPROST BACKGROUND Electronic PROtocol Submission & Tracking system • Assists the HSRO in supporting and managing all biomedical and social behavioral research • Access via standard internet browser – Internet Explorer (PC Users) – Safari (Apple Users) • Captures information capture as required by federal regulations, state laws, and institutional policies

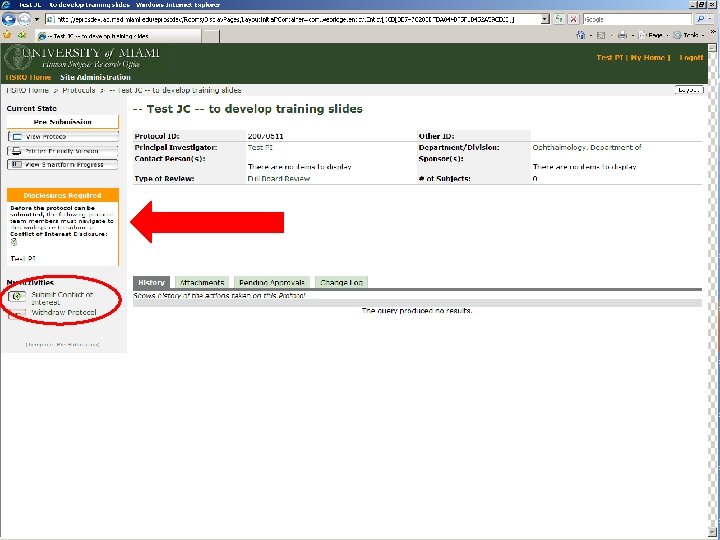
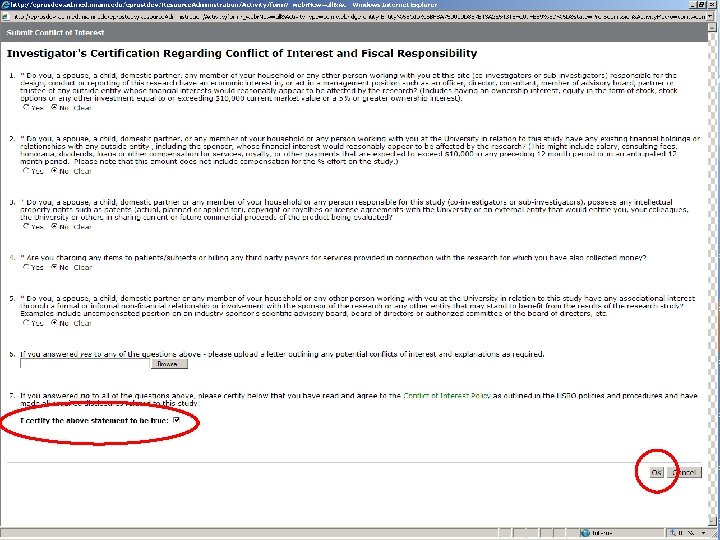
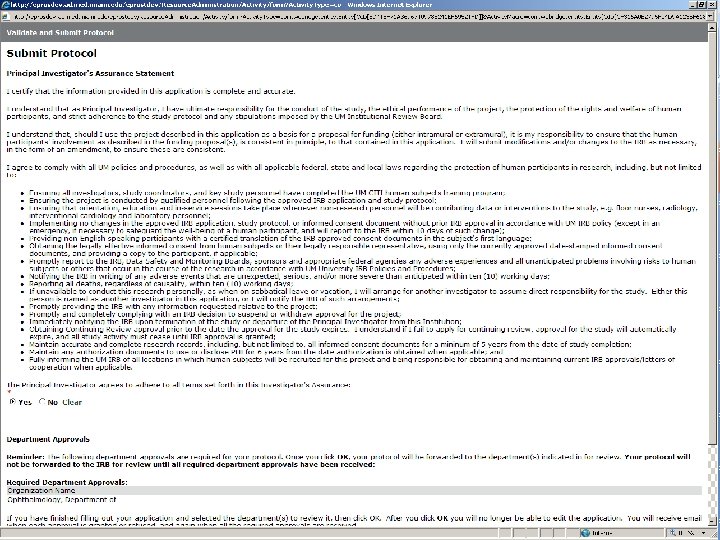
THE ELECTRONIC WORKFLOW • Electronic workflow was enabled for all Departments on Mar. 15 th, 2008 • Study teams continue to complete protocol forms in e. Prost • All members of the Protocol Team need to Submit their Conflict of Interest disclosures individually • Principal Investigator will then be able to Submit Protocol • Study will now be traveling through the workflow

BENEFITS OF WORKFLOW • Accessible anywhere you have internet access • Faster turnaround time for submissions to the HSRO • Easier tracking of studies through review process • Incorporated audit trail • Improved interaction between study teams and Institutional Review Board staff • Back-ups performed regularly by both the HSRO and Med-IT • IRB-approved and watermarked documents available online

AVAILABLE TRAINING • e. Prost User Guide available at http: //hsro. miami. edu • Up-to-date information relayed via e. Prost and HSRO e. News

SECTION 8 CREATING A STUDY IN EPROST
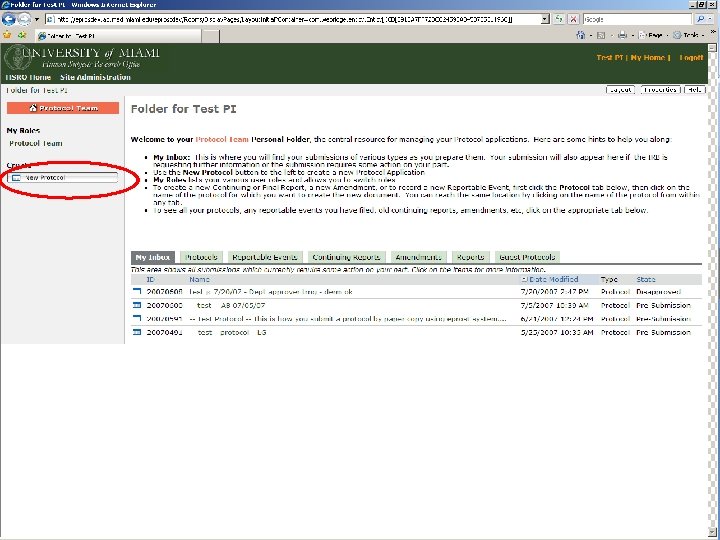
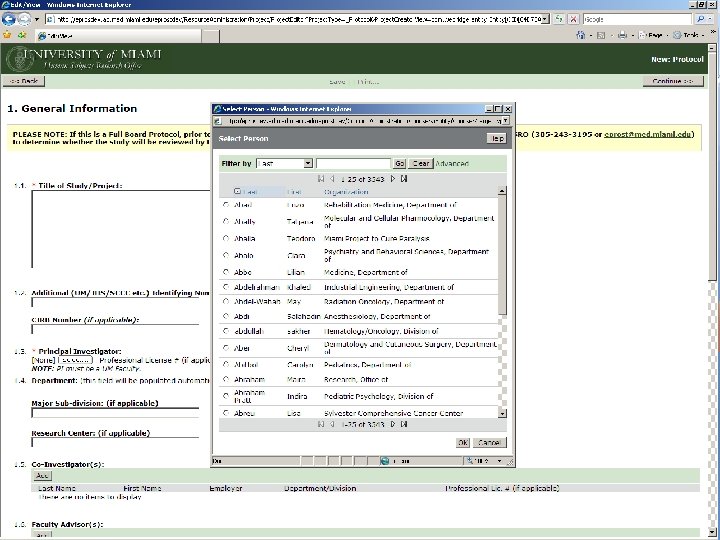
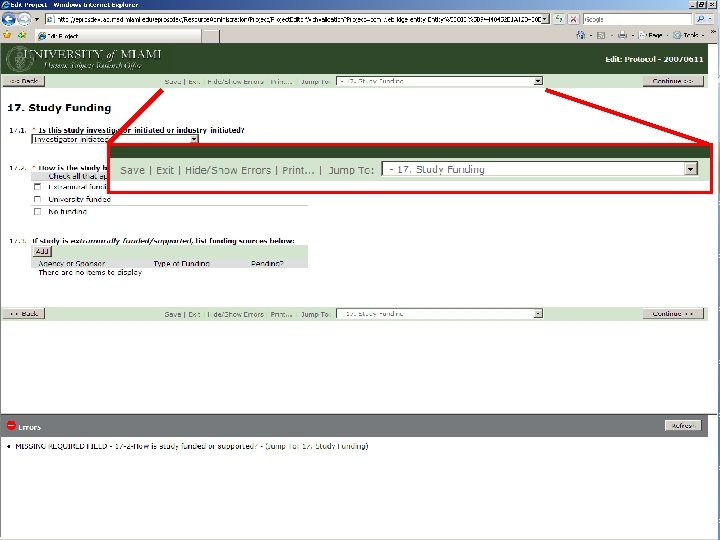
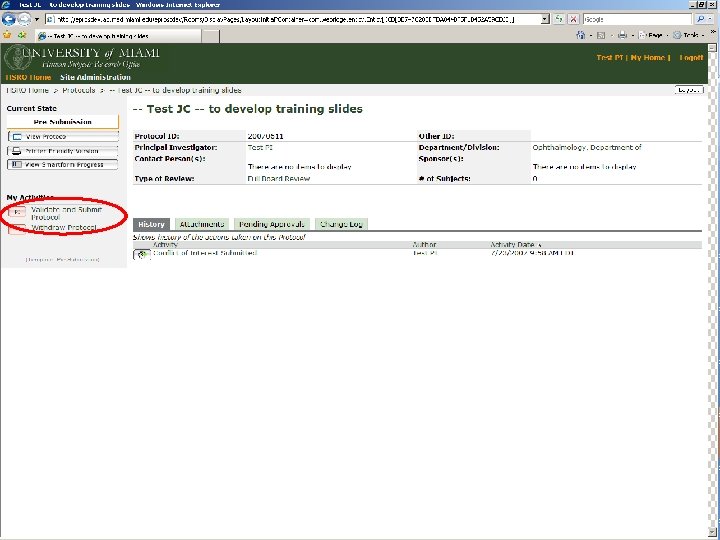
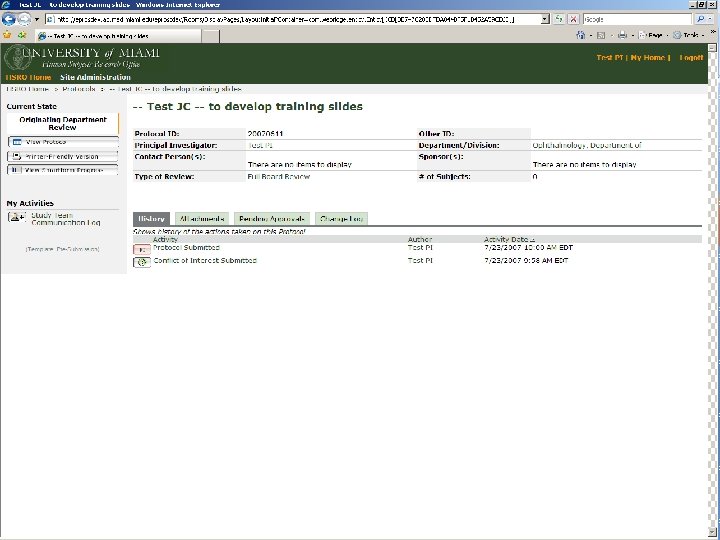

BEFORE A STUDY ARRIVES AT THE HSRO… Pre-Submission Originating Department Review Ancillary Committee Review Withdrawn Changes Required by Originating Department Changes Required by Ancillary Reviewer Disapproved Pre-Board Review

ONCE THE STUDY ARRIVES AT THE HSRO… Pre-Board Review Regulatory Review Awaiting Meeting Assignment Changes Required by Pre-Board Staff Changes Required by Regulatory Staff IRB Meeting Assigned Approved Ancillary Committee Review Expedited Review in Progress Deferred to Full Board Protocol at WIRB Exempt Review in Process Approved Deferred to Full Board

AFTER THE IRB REVIEWS THE STUDY… IRB Meeting Assigned Meeting Complete: Post. Board Review Approved Deferred to Chair Deferred to Full Board Disapproval

UM HUMAN SUBJECT RESEARCH OFFICE CONTACTS • Education – Jose Casanova, BBA, CIP • 305 -243 -3476 • Regulatory Questions / Participant Complaints – Amanda Coltes-Rojas, MPH, CIP • 305 -243 -9652 – Evelyne Bital, MA, CIP • 305 -243 -9977 • Help Desk/All Other Questions – 305 -243 -3195

- Slides: 78