Preventive measures for Thalassemia and Haemophilia Dr ZiaUlAin






 • Male gender (congenital hemophilia)")




 of these patients are born in developing and low-income")






 • INFANTS: • Age of presentation: 6 -9 mo (Hb")
 • • • BY CHILDHOOD: Growth retardation Severe anemia-cardiac dilatation")
 • Moderate pallor, usually maintains Hb >6 gm% • Anemia")
 • Usually ASYMPTOMATIC • Mild pallor, no jaundice • No")
)")





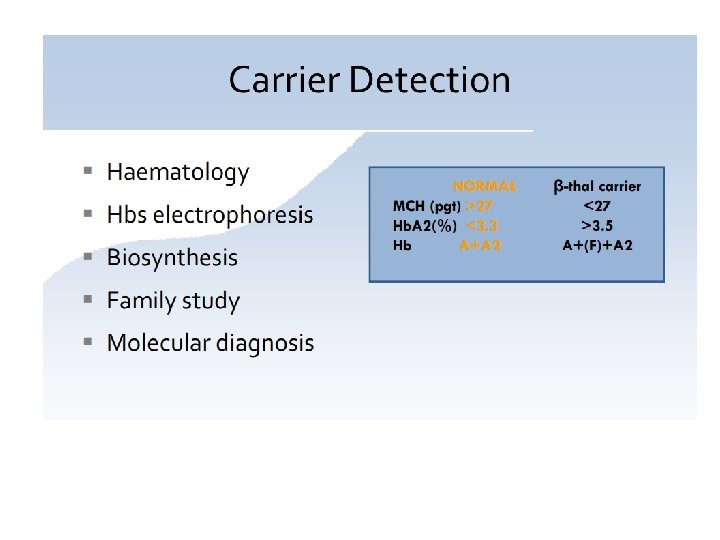
 • Population Screening(Carrier detection) • High risk group • Pregnant woman")
 • • • Risk Clinical features Patient Treatment Options Procedures")
 • • Blood samples from family members CVS biopsy/ Amniocentesis Molecular")
 CVS (1 st trimester) PGD (Pre")

- Slides: 39
Preventive measures for Thalassemia and Haemophilia Dr Zia-Ul-Ain Sabiha Assistant Professor
Learning Outcomes • Enlist the causes of thalassemia and haemophillia • Appreciate the risk factors associated with these diseases. • Learn effects of mild, moderate and severe thalassemia and haemophillia on individuals, families and communities. • Devise preventive strategies for these diseases in a community • Counsel the patients of these diseases • Case Scenarios
Haemophilia • A bleeding disorder, usually inherited, characterized by the deficiency of coagulation factor VIII or IX. • Occurs almost exclusively in males due to an X-linked pattern of inheritance. • Graded as mild, moderate, or severe, based on factor VIII or IX level. • Musculoskeletal bleeding is the most common type of hemorrhage. • Treatment consists of coagulation factor VIII or IX replacement. • A major complication of treatment is the development of inhibitory antibodies against infused factor VIII or IX.
Definition • Hemophilia is a bleeding disorder, usually inherited with an X-linked recessive inheritance pattern, which results from the deficiency of a coagulation factor. • Hemophilia A results from the deficiency of clotting factor VIII. • Hemophilia B results from the deficiency of clotting factor IX. • Acquired hemophilia is a separate noninherited condition. It is much rarer than congenital hemophilia and has an autoimmune-related etiology with no genetic inheritance pattern.
Key diagnostic factors • • History of recurrent or severe bleeding Bleeding into muscles Intracranial bleeding Prolonged bleeding following circumcision
Other diagnostic factors • Excessive bruising/hematoma • Fatigue • Menorrhagia and bleeding following surgical procedures or childbirth (female carriers) • Extensive cutaneous purpura (acquired hemophilia)
Risk factors • Family history of hemophilia (congenital hemophilia) • Male gender (congenital hemophilia) • Age >60 years (acquired hemophilia) • Autoimmune disorders, inflammatory bowel disease, diabetes, hepatitis, pregnancy, postpartum, or malignancy (acquired hemophilia)
• Because hemophilia is inherited, it cannot be prevented. • There is no cure. • Doctors can diagnose hemophilia through a blood test, typically while your child is still a fetus. • When this is done, measures can be taken to prevent bleeding, such as avoiding circumcision for baby boys and avoiding aspirin and nonsteroidal anti-inflammatory drugs (NSAIDS), which can cause bleeding. • Since carriers are usually women, genetic testing can be done to determine whether you have the gene so you can decide whether having children is right for you.
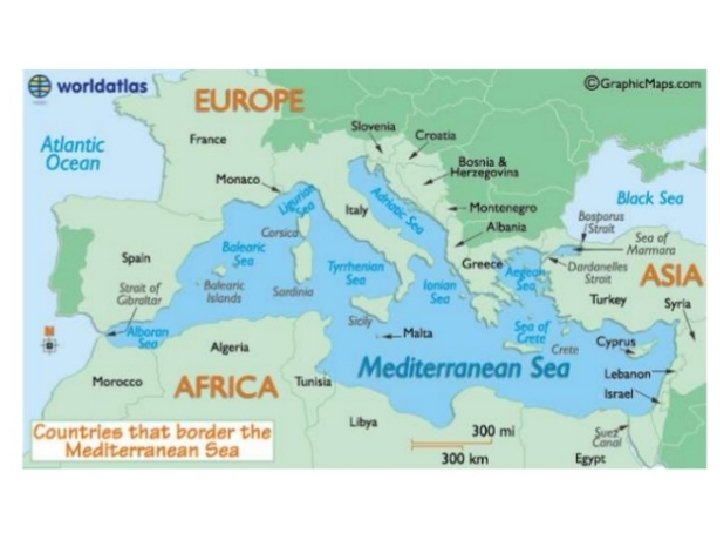
Epidemiology • Thalassemias are among the commonest autosomal recessive disorders worldwide and are prevalent in populations in the Mediterranean area, the Middle East, Transcaucasus, Central Asia, the Indian subcontinent, and the Far East. • Worldwide 56, 000 conceptions have a major thalassemia disorder of which approximately 30, 000 are affected by b-thalassemia major and 3500 succumb perinatally from the hydrops fetalis syndrome.
• Most (or many) of these patients are born in developing and low-income countries where they create an enormous health burden. • Together with sickle cell anemia, it has also been estimated that, worldwide, 9 million carriers become pregnant annually and 1. 33 million pregnancies are at risk for a thalassemia major condition
Introduction • β-Thalassaemia major is an inherited blood disorder presenting with anaemia at 4 – 6 months of age.
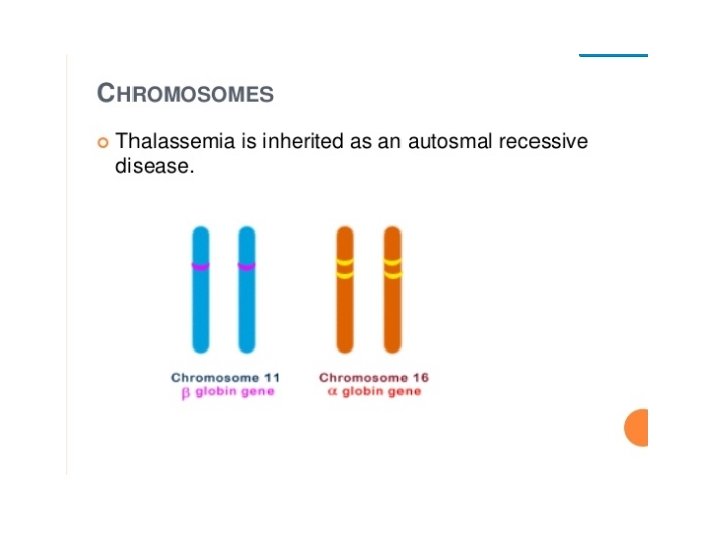
INHERITANCE • • Autosomal recessive • Beta thal - point mutations on chromosome 11 • Alpha thal - gene deletions on chromosome 16
BASICS - 3 types of Hb 1. Hb A - 2 α and 2 β chains forming a tetramer • 97% adult Hb • Postnatal life Hb A replaces Hb F by 6 months 2. Fetal Hb – 2α and 2γ(gamma) chains • 1% of adult Hb • 70 -90% at term. Falls to 25% by 1 st month and progressively 3. Hb A 2 – Consists of 2 α and 2 δ (delta)chains • 1. 5 – 3. 0% of adult Hb
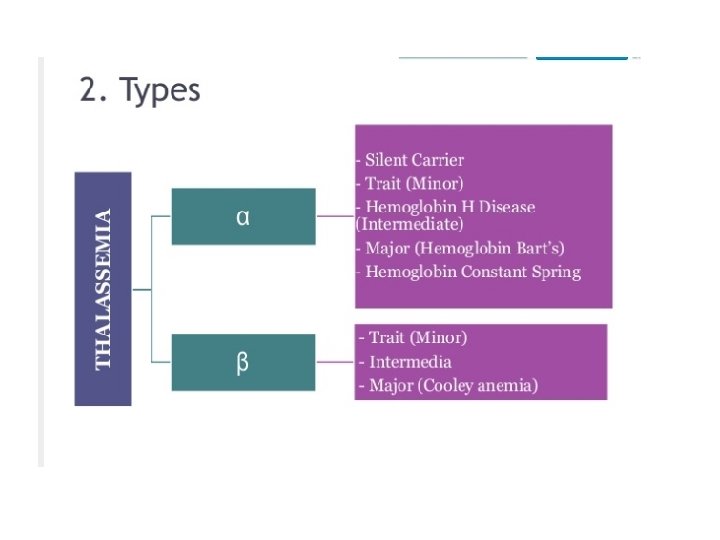
Classification • If synthesis of α chain is suppressed – level of all 3 normal Hb A (2α , 2β), A 2 (2α , 2 δ), F(2α , 2γ) reduced – alpha thalassemia • If β chain is suppressed - adult Hb is suppressed - beta thalassemia
CLASSIFICATION OF β THALASSEMIA CLASSIFICATION GENOTYPE CLINICAL SEVERITY β thal minor/trait β/β+, β/β 0 Silent β thal intermedia β+ /β+, β+/β 0 Moderate β thal major β 0/ β 0 Severe
CLINICAL FEATURES (THAL MAJOR) • INFANTS: • Age of presentation: 6 -9 mo (Hb F replaced by Hb A) • Progressive pallor and jaundice • Cardiac failure • Failure to thrive, gross motor delay • Feeding problems • Bouts of fever and diarrhea • Hepatospleenomegaly
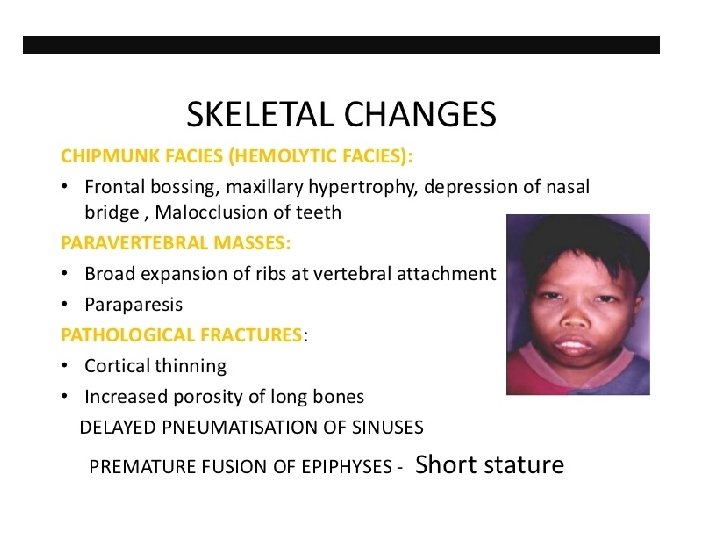
CLINICAL FEATURES (THAL MAJOR) • • • BY CHILDHOOD: Growth retardation Severe anemia-cardiac dilatation Transfusion dependant Changes in skeletal system
CLINICAL FEATURES (THAL INTERMEDIA) • Moderate pallor, usually maintains Hb >6 gm% • Anemia worsens with pregnancy and infections (erythroid stress) • Less transfusion dependant • Skeletal changes present, progressive spleenomegaly • Growth retardation • Longer survival than Thal major
CLINICAL FEATURES (THAL MINOR) • Usually ASYMPTOMATIC • Mild pallor, no jaundice • No growth retardation, no skeletal abnormalities, no spleenomegaly • MAY PRESENT AS IRON DEFICIENCY ANEMIA (Hypochromic microcytic anemia) • Unresponsive/ refractory to Fe therapy • Normal life expectancy
Treatment Repeated Blood transfusion ( at 4 -6 wks interval (Hb~ 9. 5 gm/dl)) (PRBCs) Hb to be maintained – • Hypertransfusion : >10 gm/dl • Supertransfusion : >12 gm/dl – Iron overload ( iron Chelation therapy) Bone marrow transplant( best method for cure) Spleenectomy (5 -6 years of age)
Treatment GENE MANIPULATION AND REPLACEMENT • Remove defective β gene and stimulate γ gene • 5 -azacytidine increases γ gene synthesis • Hb F AUGEMENTATION • Hydroxyurea • Myelaran • Butyrate derivatives • Erythropoetin in Thal intermedia
OTHER SUPPORTIVE MEASURES • Tea – thebaine and tannins– chelate iron • Vitamin C – increases iron excretion • Restrict Fe intake – decrease meat, liver, spinach • Folate – 1 mg/day • Genetic counseling • Psychological support • Hormonal therapy – GH, estrogen, testosterone, L -thyroxine • Treatment of CCF
Prognosis • Life expectancy: 15 -25 yrs • Untreated: < 5 yrs
Thalassemia Control Programs Prevention • • Public education Carrier screening Genetic Counseling Prenatal diagnosis
Public Education • • • Schools Leaflets Media Conferences/ Seminar Professionals To inform NOT to stigmatize
Prevention Program(Carrier Screening) • Population Screening(Carrier detection) • High risk group • Pregnant woman
Prevention program (Genetic Counseling) • • • Risk Clinical features Patient Treatment Options Procedures to follow
Prevention Program(prenatal diagnosis) • • Blood samples from family members CVS biopsy/ Amniocentesis Molecular analysis(ARMs sequencing etc) Diagnosis
Prenatal Diagnosis • • Amniocentesis( 2 nd trimester) CVS (1 st trimester) PGD (Pre implantation genetic diagnosis) Non Invasive prenatal diagnosis
• Thank you