PLANNING AND PERFORMING A RANDOMIZED CONTROLLED CLINICAL TRIAL


























 that impact study outcome equally distributed in study")
 • Assures equal number")













 vs. approved therapy (S) – Outcome:")
 to –")



 vs. drug (β-blocker) for prevention")













")










 • Prevent selective")


 – Controlled")















 •")


- Slides: 94
PLANNING AND PERFORMING A RANDOMIZED CONTROLLED CLINICAL TRIAL
REPRODUCIBILITY IN RESEARCH • “Growing alarm about results that cannot be reproduced” Nature Supplement, Challenges in Irreproducible Research, October 7, 2015 • “Reproducibility, rigor, transparency and independent verification are cornerstones of the scientific method” NIH-Science-Nature Workshop on Reproducibility and Rigor of Preclinical Research Nature 2014; 515: 7
ENHANCING REPRODUCIBILITY AND RIGOR IN CLINICAL RESEARCH • Study design of high methodologic quality – Minimizes bias: better estimate of “truth”
ENHANCING REPRODUCIBILITY AND RIGOR IN CLINICAL RESEARCH • Study design of high methodologic quality – Minimizes bias: better estimate of “truth” • Transparent (full and clear) presentation of methods and analyses – Enables assessment of methods and results – Allows duplication of study, re-analysis of results
ENHANCING REPRODUCIBILITY AND RIGOR IN CLINICAL RESEARCH • Study design of high methodologic quality – Minimizes bias: better estimate of “truth” • Transparent (full and clear) presentation of methods and analyses – Enables assessment of methods and results – Allows duplication of study, re-analysis of results • Registration before trial begins – Prevents changing design or pre-specified outcomes/analyses without explanation
ENHANCING REPRODUCIBILITY AND RIGOR IN CLINICAL RESEARCH • Study design of high methodologic quality – Minimizes bias: better estimate of “truth” • Transparent (full and clear) presentation of methods and analyses – Enables assessment of methods and results – Allows duplication of study, re-analysis of results • Registration before trial begins – Prevents changing design or pre-specified outcomes/analyses without explanation • Trials reported per international standards – All appropriate elements included
REPRODUCIBILITY ISSUES Preclinical Research Most Susceptible • Clinical trials seem to be less at risk because already governed by regulations that stipulate rigorous design and independent oversight (randomization, blinding, power estimates, pre-registration in standardized, public databases, oversight by IRBs and DSMBs) and adoption of standard reporting elements Collins FS, Tabak LA. Nature 2014; 505: 612
CLINICAL TRIALS
LEVELS OF EVIDENCE FOR CLINICAL RESEARCH STUDY DESIGNS Schillaci et al. Hypertension. 2013; 62: 470
OBSERVATIONAL VS. RANDOMIZED TRIALS
OBSERVATIONAL VS. RANDOMIZED TRIALS • “There are knowns; there are things we know. We also know there are known unknowns; that is to say, we know there are some things we do not know. But there also unknowns--the ones we don’t know. ” - Donald Rumsfeld
OBSERVATIONAL VS. RANDOMIZED TRIALS • Observational Studies – Distribution of baseline factors that may impact outcome (e. g. , age, meds, comorbidities) vary in study groups • Known knowns: known to impact outcome, collected – Statistical adjustment, matching • Known unknowns: known to impact outcome, can’t be collected • Unknown unknowns: don’t know impact outcome, not collected • Randomized Trials – All factors (known and unknown) that may impact outcome equally distributed among study groups
RANDOMIZED CONTROLLED TRIAL • Only randomized trials of sufficient size can adequately control for known and unknown confounding variables to minimize bias • No substantive differences between groups except study intervention (randomly assigned) – Difference between groups in predefined outcome can be attributed to the intervention being studied Hennekens & Buring Epidemiology in Medicine. 1987
DO WE ALWAYS NEED AN RCT TO DOCUMENT BENEFIT OF AN INTERVENTION?
DO WE ALWAYS NEED AN RCT TO DOCUMENT BENEFIT OF AN INTERVENTION? • “Perception that parachutes are a successful intervention based largely on anecdotal evidence” • No RCTs identified in systematic review – Under exceptional circumstances apply common sense BMJ 2003; 327: 1459
RCT MAY NOT BE POSSIBLE OR PRACTICAL • Not ethical/possible to assign intervention – Cigarette smoking and lung cancer – H. pylori infection and ulcers • Impractically large sample size – Very low-incidence outcome • e. g. , rare side effect of medication • Impractically long duration – Outcome requires many years to develop • e. g. , development of cancer
RANDOMIZED CONTROLLED TRIALS First Steps
RANDOMIZED CONTROLLED TRIALS First Steps • Clinically relevant question – Greatest impact if limited information or high variability in care or outcomes – Can be answered by properly designed RCT • Feasible to perform at your center(s)
RANDOMIZED CONTROLLED TRIALS First Steps • Clinically relevant question – Greatest impact if limited information or high variability in care or outcomes – Can be answered by properly designed RCT • Feasible to perform at your center(s) • Systematic review – Identify available information – Justify importance of question – Help design study
RANDOMIZED CONTROLLED TRIALS First Steps • Define key elements of study – Population – Intervention – Comparator – Outcome • State primary hypothesis • Expected result for primary outcome in population – e. g. , in patients with cirrhosis fewer deaths with new intervention vs. control
STUDY DESIGN
RANDOMIZATION
RANDOMIZATION • Generate sequence of allocation – Computer generated, random numbers table – Randomize in blocks • Other features of randomization include – Concealed allocation – Non-manipulable allocation schedule • Off-site randomization schedule ideal – Stratification • Most important factor(s) that may impact endpoint
CONCEALED ALLOCATION • Concealed allocation is an extension of randomization • When obtaining informed consent to enroll a patient into a trial, the investigator does not know if the next patient will get new treatment or control
CONCEALED ALLOCATION • RCT comparing new therapy vs. placebo for abdominal pain in irritable bowel syndrome • Investigator interviews the next eligible patient, who complains of long-term severe, unrelenting symptoms that have never responded to previous medical therapy • Next patient to enter trial will get placebo
CONCEALED ALLOCATION • Investigator thinks that placebo is unlikely to relieve abdominal pain in this patient – Investigator may subconsciously try to convince patient not to enroll in the trial • Consequence: patients with severe abdominal pain will NOT be evenly divided between new therapy and placebo groups
STRATIFICATION • To assure baseline factor(s) that impact study outcome equally distributed in study groups – Especially useful in smaller trials • Choose factor(s) that have greatest impact on primary outcome – Aspirin use in MI study • Separate randomization schedules for patients with and without factor
RANDOMIZATION Block Size (e. g. , 4, 10, 20; Random) • Assures equal number in each study arm for every successive block of patients enrolled – Prevents unequal numbers in study arms – Prevents differences in distribution over time • e. g. , study intervention mostly early, comparator mostly later – Disadvantage: if block size figured out, next allocation may be predictable (unconcealed)— selection bias • Larger block sizes; random sequences of block sizes
BLINDING
BLINDING • Not known if subject getting new therapy or control – Subjects – Healthcare providers making management decisions – Investigators collecting/analyzing data • Prevents bias in management decisions and in assessment of outcomes by subject or investigator – Knowledge receiving placebo or active drug may influence • Administration of anotherapy that my impact outcome • Assessment of symptoms, signs (endpoints)
BLINDING • Identical appearing therapies – Real vs. sham surgery/procedure • Surgical team uninvolved in further care/assessment • Double-dummy – Subjects receive identical active and control therapy together • Side effect of a therapy may unblind subjects – Assess whether unblinded
TREATMENT EFFECT OVERESTIMATED WITHOUT RANDOMIZATION AND BLINDING 35 30 20 ase 0 Not Randomized Treatment Placebo Treatment 5 Placebo 10 Treatment 15 Placebo Fatality Rates 25 Concealed Allocation; Blinded Chalmers, et al. N Engl J Med 1983; 309: 1358
PATIENT POPULATION • Inclusion and exclusion criteria – Broad: exclude few, more generalizable – Restricted: exclude many, less generalizable • Prospectively screen consecutive patients with condition of interest – Skipping patients may introduce bias • Screening log – Subjects screened, but not enrolled • Brief characteristics, reason not enrolled – ? Differences from those enrolled – Is study generalizable?
STUDY INTERVENTIONS • Define all aspects of study interventions so uniform in trial, able to be reproduced • Control – Placebo control • Best to define efficacy of study therapy • May not be ethical, practical – Can’t withhold standard care if documented effective – Active control (a current standard) • Hypothesis: new therapy superior, non-inferior, or equivalent to active control
ENDPOINTS • What do you want to achieve with the new intervention – Primary endpoint – Additional endpoints – Surrogate vs. clinical endpoints • Surrogate endpoint: measure of treatment effect felt likely to correlate with clinical endpoint • e. g. , gastric acid inhibition for ulcer prevention
CLINICALLY MEANINGFUL ENDPOINTS PREFERRED • Which study endpoint would alter practice? – Lab test (CRP) or clinical outcome (death) • Studies of intermediate/surrogate endpoints may indicate areas for further research, but generally don’t alter patient management • Some surrogate endpoints are accepted as “true” indicators of clinical outcomes – e. g. , blood pressure, cholesterol, colon polyps
SAMPLE SIZE DETERMINATION
SAMPLE SIZE DETERMINATION Assumptions for Superiority Study • Primary endpoint result for the intervention • Primary endpoint result for the comparator – Assumptions based on available data, clinical judgment • Hypothesized difference should be clinically meaningful, realistic • α – p = 0. 05 – probability of finding difference when doesn’t exist (type I) • Power (1 – β) – Probability of finding difference when does exist • e. g. , 80%, 90% • β: probability of not finding a difference when does exist (type II)
SAMPLE SIZE DETERMINATION Why Did They Stop the Study When They Did? • RCT: Wonderdrug vs. placebo in pancreatic cancer • Primary endpoint: 5 -yr survival – Wonderdrug: 50% – Placebo: 10% – P-value (α = 0. 05) – 90% power to detect 40% difference between Wonderdrug and placebo • 52 patients required (if 1: 1 randomization)
SAMPLE SIZE DETERMINATION Assumptions for Non-Inferiority Study • Determine non-inferiority margin – Clinical: maximal difference that would be considered clinically non-inferior • Not unacceptably worse than the control – Statistical: maintain benefit above placebo • Control is 20% more efficacious than placebo • Margin of 10% retains half control treatment effect • Margin (e. g. , control – test drug = 3%) less than upper bound of CI of difference observed in study – Difference = 0% (95% CI -5% to 5%): Not non-inferior – Difference = 0% (95% CI -1% to 1%): Non-inferior
SAMPLE SIZE DETERMINATION Non-Inferiority Study • Determine non-inferiority margin – Clinical: maximal difference that would be considered clinically non-inferior Potential reasons toworse do non-inferiority • Not unacceptably than the control study • New interventionmaintain has some benefit other advantage that would recommend it – Statistical: above placebo • Control is (non-inferior) 20% more efficacious than placebo if efficacy similar to current standard therapy • Margin of safer, 10% retains half effect • e. g. , cheaper, easier to usecontrol (pill vs. treatment enema), more readily • Margin (e. g. , control – test drug = 3%) less than available (oral rehydration vs. IV fluids); commercial upper bound of CI of difference observed in study – Difference = 0% (95% CI -5% to 5%): Not non-inferior – Difference = 0% (95% CI -1% to 1%): Non-inferior
NON-INFERIORITY STUDY: FDA EXAMPLE • New thrombolytic (R) vs. approved therapy (S) – Outcome: Mortality • New thrombolytic must retain ≥ 50% benefit of approved therapy to be acceptable alternative • Mortality difference S vs. placebo – 2. 6% (lower bound 95% CI = 2. 1%) • Study has to rule out 1. 05% increase in mortality with R compared to S – 95% CI of difference in mortality for R vs. S < 1. 05% • Accept 1. 05% increase as not unacceptably worse http: //www. fda. gov/downloads/Drugs/Guidance. Compliance. Regulatory. Information/Guidances/UCM 202140. pdf
IS THE SAMPLE SIZE FEASIBLE • Review medical records at study center(s) to – Determine number who meet enrollment criteria – Confirm assumptions about outcomes • “Preparatory to research” review doesn’t require IRB approval of the protocol – “This type of access is limited to a review of data to assist in formulating a hypothesis, determining the feasibility of conducting the study. . . or other similar uses that precede the development of an actual protocol. ” – Submit Request for Access form
POPULATIONS FOR ANALYSIS
POPULATIONS FOR ANALYSIS Intention to Treat Analysis All randomized patients are included in final data analysis Per Protocol Analysis Only patients who complete the trial according to protocol are analyzed 45
POPULATIONS FOR ANALYSIS • Intention-to-treat population – All patients randomized regardless of follow-up or receipt of study intervention • Per-protocol population excludes those who – Did not receive sufficient study intervention – Did not return for adequate follow-up – Had major violations of inclusion criteria • e. g. , did not have the disease being studied – Had major violations during the study • Took non-study PPI during PPI vs. placebo study
INTENTION-TO-TREAT ANALYSIS Example • Comparison of radiology procedure (TIPS) vs. drug (β-blocker) for prevention of recurrent variceal bleeding with death as the primary endpoint • If a patient is randomized to get TIPS and dies from bleeding before the procedure can be done, should the patient be included in the final data analysis?
POPULATIONS FOR ANALYSIS • Choose the most conservative analysis – Less likely to favor intervention, be overly optimistic • Superiority study – Per-protocol assesses intervention under optimal circumstances (not real world, ignores study quality) • e. g. , excluded if non-adherence, protocol violations, drop-out – ITT avoids bias to treatment difference and superiority • Non-inferiority study – ITT can bias to no treatment difference (non-inferiority) • e. g. , non-adherence, drop-outs, misclassified subjects/endpoints – Per protocol analysis should be included
COMPLETE FOLLOW-UP OF PATIENTS Another Requirement for High Methodologic Quality • If numerous patients are lost to follow-up, results of the trial may not be accurate – Predefine method to deal with such patients • Last observation carried forward • Imputation methods • Re-calculate results assuming that patients lost to follow-up in new treatment group had bad outcome and patients lost to follow-up in control group had good outcome
RECRUITMENT AND RETENTION • Engagement, communication with participants before and throughout trial – Brochures, ads, social media, phone/text/email/websites • Reminders for study personnel and participants • Benefits of participation for subjects – Societal, personal, financial reimbursement for time • Benefits of participation for research personnel – Academic (e. g. , authorship), financial • Identify and minimize barriers to participation – Easy access to study personnel and activities • Participation as non-onerous as possible
STUDY ANALYSIS
STUDY ANALYSIS • Predefine presentation of data – Proportions vs. time-to-event curves – Mean vs. median • Predefine statistical analyses – Comparisons for primary, additional outcomes – Subgroup analyses – Other analyses • e. g. , multivariable analyses, sensitivity analyses
COMPARING THE STUDY GROUPS
P-VALUE: DID DIFFERENCE BETWEEN TREATMENT AND CONTROL OCCUR DUE TO CHANCE? • Null hypothesis – “True” proportion of success with treatment equals “true” proportion of success with control • If the null hypothesis correct and treatments are equally effective, p-value indicates – Probability of observing a difference between treatment and control at least this large • Probability that difference at least this large is due to chance
P-VALUE: DID DIFFERENCE BETWEEN TREATMENT AND CONTROL OCCUR DUE TO CHANCE? • A small p-value (< 0. 05) means finding a difference at least this large is unlikely if the null hypothesis (treatments equally effective) is true – Reject the null hypothesis
P-VALUE WITH MULTIPLE COMPARISONS • Increased chance comparisons “significant” • Type 1 error: 2 endpoints: ~10%; 10 endpoints: ~40% – Process to control rate of false positives • Single primary outcome, limited additional outcomes • Correct for multiplicity – Bonferroni (p/# analyses: 0. 05/10 = 0. 005) – Hierarchical testing (predefined sequence, stop when p>0. 05) – Other techniques • Provide results without claims of “significance” – Don’t perform many analyses stating significance is p<0. 05 for all analyses http: //www. fda. gov/downloads/Drugs/Guidance. Compliance. Regulatory. Information/Guidances/UCM 536750. pdf
95% CONFIDENCE INTERVALS A More Precise Tool to Assess the Results • New therapy vs. control – Absolute difference = 9%, 95% CI 4 – 14% – If trial repeated 100 times, difference would be between 4% and 14% in 95 of 100 trials
95% CONFIDENCE INTERVALS A More Precise Tool to Assess the Results • Tighter the interval around the observed result, the more precise the estimate – Difference = 0%: -30% to 30% vs. -1% to 1%
95% CONFIDENCE INTERVALS A More Precise Tool to Assess the Results • Tighter the interval around the observed result, the more precise the estimate – Difference = 0%: -30% to 30% vs. -1% to 1% • If bounds of 95% CI – Both >0 (for absolute difference) or >1 (for RR) of new therapy vs. control • Consistent with new treatment more efficacious – Difference=20%, 10 -20%; RR=2. 5, 1. 5 -3. 5
95% CONFIDENCE INTERVALS A More Precise Tool to Assess the Results • Tighter the interval around the observed result, the more precise the estimate – Difference = 0%: -30% to 30% vs. -1% to 1% • If bounds of 95% CI – Both >0 (for absolute difference) or >1 (for RR) of new therapy vs. control • Consistent with new treatment more efficacious – Difference=20%, 10 -20%; RR=2. 5, 1. 5 -3. 5 – Cross 0 (for absolute difference) or 1 (for RR) • Consistent with new treatment better or worse – Difference=5%, -5% to 15%; RR=1. 2, 0. 6 to 2. 0
95% CONFIDENCE INTERVALS A More Precise Tool to Assess the Results Success (Small Study) 13/26 (50%) Success (Large Study) 1000/2000 (50%) Control 10/26 (38%) 973/2000 (48. 7%) p-value 0. 40 Difference 12% 1. 3% -15% to 38% -1. 8% to 4. 4% Large effect size; high uncertainty Small effect size; low uncertainty Treatment 95% CI Interpretation Connor. AJG 2004; 99: 1638
SUBGROUP ANALYSES • Does intervention behave differently in subset of population – e. g. , men vs. women, old vs. young • Should be pre-defined and justified – Post hoc more risk of bias since results known • Less power than overall analysis – More analyses increases chance of significance occurring by chance – Smaller number in each subgroup
SUBGROUP ANALYSES • Don’t assess if treatment effect is significant in each subgroup – Don’t compare outcome for new therapy vs. control in each subgroup • If baseline assumptions about results for outcomes in sample size calculation were correct, wouldn’t expect smaller subgroups to show significant difference even if treatment effect exactly the same as in overall group
SUBGROUP ANALYSES • Don’t assess if treatment effect is significant in each subgroup Study of New Therapy vs. Control – Don’t compare outcome for new therapy vs. control • in each subgroup 200 subjects to show 20% difference (50% vs. 30%) • Overall: assumptions 50/100 (50%) vs. 30/100 (30%) for p=0. 01 • If baseline about results • Men: in sample 20/40 size (50%) vs. 12/40 (30%) outcomes calculation were p=0. 11 correct, wouldn’t 30/60 expect smaller subgroups to • Women: (50%) vs. 18/60 (30%) p=0. 04 show significant difference even if treatment effect exactly the same as in overall group
SUBGROUP ANALYSES Compare Treatment Effects in Subgroups • Is treatment effect (e. g. , RR of death for new therapy vs. control) different for old vs. young? – Is there a treatment by subgroup interaction? – Qualitative interaction • Treatment effects in opposite direction – e. g. , RR < 1 for old; RR > 1 for young – Quantitative interaction • Treatment effects of different magnitude--statistically heterogeneous (interaction test p < 0. 05) • e. g. , old: RR = 0. 4, 0. 1 -1. 0; young: RR = 0. 9, 0. 7 -1. 2
DATA SAFETY MONITORING BOARD
DATA SAFETY MONITORING BOARD • Independent, external experts – No involvement, conflicts related to study • Periodic assessments – Data quality, timeliness; trial site performance – Recruitment, accrual and retention – Safety and efficacy outcomes (risk-benefit) – Factors external to the study • New data may impact safety or ethics of study • Make recommendations to continue, modify, or terminate study
DATA SAFETY MONITORING BOARD • Independent, external experts – No involvement, conflicts related to study Periodic assessments • • “Ensures that a clinical trial is stopped if the – Data quality, timeliness; trial site performance benefit-risk balance or the – Recruitment, accrual for and participants retention – Safety and efficacy outcomes expected value to society no(risk-benefit) longer justifies – Factors external to the study • New data may impact safety or ethics. Lewis of study continuing” et al. JAMA 2016; 316: 2359 • Make recommendations to continue, modify, or terminate study
INTERIM ANALYSIS Predefined Formal Data Analysis Partway Through Trial • Efficacy: outcome assumptions used in sample size determination may be inexact/uncertain – Modify study (adaptive design) – Stop study (may have predefined stopping rules) • Futility: no possibility can document benefit • Efficacy: unequivocal benefit in clinically important outcome – Very conservative p-value (e. g. , 0. 001) • Prevents incorrect conclusion (vs. p-value closer to 0. 05) • Minimal α spending with multiple comparisons • Final significant p-value only slightly below 0. 05
INTERIM ANALYSIS Predefined Formal Data Analysis Partway Through Trial • Efficacy: outcome assumptions used in sample size determination may be inexact/uncertain – Modify study (adaptive design) – Stop study (may have predefined stopping rules) • Futility: no possibility can document benefit • Efficacy: unequivocal benefit in clinically important outcome – Very conservative p-value (e. g. , 0. 001) • Prevents incorrect conclusion (vs. p-value closer to 0. 05) • Minimal α spending with multiple comparisons • Final significant p-value only slightly below 0. 05 • Safety: Concern/uncertainty about therapy’s safety – Stop study if unacceptable risk-benefit balance – Add additional monitoring, therapies, safeguards
TRIAL REGISTRATION
PURPOSE OF TRIAL REGISTRATION International Committee of Medical Journal Editors (ICMJE) • Prevent selective publication and selective reporting of research outcomes • Prevent unnecessary duplication of research • Help public know of planned or ongoing trials into which they might want to enroll • Give ethics review boards considering approval of new studies a view of similar work and data relevant to the research http: //icmje. org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration. html
CLINICAL TRIAL DEFINITION • Clinical. Trials. gov – Participants receive intervention per protocol • Drug, device, procedure, diet, brochure, video – Outcomes measured to determine safety, efficacy • ICMJE (Medical Journal Editors) – Prospectively assigns people to an intervention, with or without concurrent comparison, to study the cause-andeffect relationship between a health-related intervention and a health outcome • NIH – Human subject(s) prospectively assigned to intervention to evaluate the effects on health-related biomedical or behavioral outcomes https: //clinicaltrials. gov/ct 2/about-studies/learn; http: //icmje. org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration. html; https: //oir. nih. gov/sourcebook/intramural-program-oversight/intramural-data-sharing/guide-fdaaa-reporting-research-results/nih-definition-clinical-trial
CLINICAL TRIAL DEFINITION • Clinical. Trials. gov – Participants receive intervention per protocol • Drug, device, procedure, diet, brochure, video – Outcomes measured to determine safety, efficacy Clinical Trial • ICMJE (Medical Journal Editors) – Prospectively assigns people to an intervention, with or • Prospective without concurrent comparison, to study the cause-and • Assignment of intervention effect relationship between a health-related intervention and a health outcome • Measurement of health outcomes after intervention • NIH – Human subject(s) prospectively assigned to intervention to evaluate the effects on health-related biomedical or behavioral outcomes https: //clinicaltrials. gov/ct 2/about-studies/learn; http: //icmje. org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration. html; https: //oir. nih. gov/sourcebook/intramural-program-oversight/intramural-data-sharing/guide-fdaaa-reporting-research-results/nih-definition-clinical-trial
WHY REGISTER A CLINICAL TRIAL • Required by law (U. S. FDA) – Controlled clinical investigations of FDA-regulated drug, biologic, or device other than Phase 1 (drugs/biologics) or small feasibility studies • Enable publication (ICMJE) – All interventional studies, including Phase 1 • Ethical (e. g. , WHO, Declaration of Helsinki) – “The registration of all interventional trials is a scientific, ethical and moral responsibility” (WHO) https: //clinicaltrials. gov/ct 2/manage-recs/background
REPORTING TRIALS
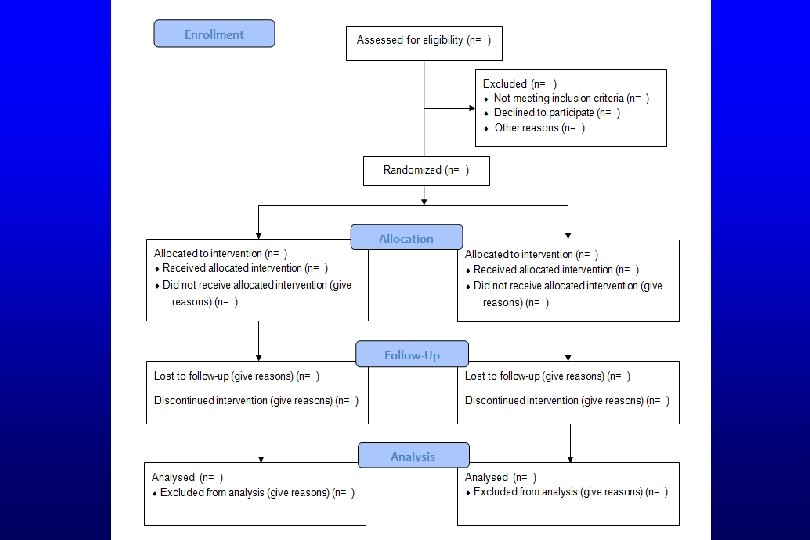
GUIDELINES FOR REPORTING TRIALS • Ensures all important elements of study are included and reported appropriately • CONSORT guidelines – Framework for reporting RCTs – Required by many journals – Checklist for the content of the title, abstract, introduction, methods, results, and discussion – CONSORT flow diagram CONSORT=Consolidated Standards of Reporting Trials
RANDOMIZED CONTROLLED TRIALS • Best design to minimize bias in assessment of an intervention – Factors to provide high methodologic quality • e. g. , concealed allocation, blinding, complete f/u • Predefined outcomes, sample size, analyses • Pre-trial registration ensures no change from original design without explanation • Reporting guidelines ensure all important elements provided in publication
ASSESSMENT OF RCT
BACKGROUND AND HYPOTHESIS • Background – Relevance, importance, novelty of topic – Rationale clear • Hypothesis – Primary hypothesis stated
BACKGROUND AND HYPOTHESIS • Background – Colonoscopy with sedation extremely common – Guidelines suggest diphenhydramine if difficult to sedate with standard benzodiazepine/opioid • No studies assessed this practice • Hypothesis – Introduction • Goal is to determine if diphenhydramine superior to continued midazolam in difficult-to-sedate patients – Statistics • Hypothesis: diphenhydramine is superior in achieving adequate moderate sedation in difficult-to-sedate patients
TRIAL REGISTRATION AND REPORTING • Trial Registered – e. g. , Clinicaltrials. gov • Trial reported per guidelines – CONSORT for RCTs
TRIAL REGISTRATION AND REPORTING • Trial Registered – Methods • Registered with Clinical. Trials. gov (NCT 01769586) • Trial reported per guidelines – CONSORT flow diagram (Figure 1) – No other mention of CONSORT • Journal requires CONSORT checklist with submission
STUDY DESIGN • Randomization – Method of randomization stated – Allocation concealed – Block size – Stratification • Blinding – Patients, caregivers, investigators – Method of blinding stated (e. g. , double-dummy)
STUDY DESIGN • Randomization – Computer generated by uninvolved individual – Allocation concealed (opaque covering) – Block size: not stated – Stratification: Not stated (presume not done) • Blinding – Patients, caregivers, investigators blinded • Person giving drug unblinded (not otherwise involved) – Better if coded syringes from central location – Method of blinding: identical syringes
STUDY DESIGN • Population – Inclusion, exclusion criteria clearly stated • Broad vs. restrictive – Consecutive patients enrolled • Intervention and Control – Characterized fully – Clinically appropriate • Simulate standard practice • Control is an acceptable standard of care
STUDY DESIGN • Population – Inclusion, exclusion criteria clear • Broad in that virtually anyone undergoing colonoscopy who was difficult to sedate – Consecutive patients enrolled • Yes, but only days when 2 investigators available • Intervention and Control – Characterized fully: Yes – Clinically appropriate • Simulate standard practice and control acceptable standard of care: Yes
STUDY DESIGN • Primary and additional outcomes defined – Clinically relevant – Appropriately measured • Sample size assumptions, calculation – Superiority vs. non-inferiority study – Assumptions reasonable
STUDY DESIGN • Primary and additional outcomes defined – Adequate sedation is clinically relevant • Patient and endoscopist assessment most relevant? – All patients could receive more meds if not adequately sedated – MOAA/S valid instrument to measure sedation • ? difference in MOAA/S performance with different drugs • Sample size assumptions, calculation – Superiority study – Outcome assumptions not well justified • Based on “clinical experience” • 20% difference reasonable--clinically meaningful
ANALYSES • Population – ITT vs. per-protocol – Full accounting of subjects • Primary and additional analyses – Predefined analyses presented • • Appropriate statistical comparisons Magnitudes and precision of effect Subgroup analyses (choice of subgroups justified) Other analyses: e. g. , sensitivity, multivariable – Post hoc analyses
ANALYSES • Population – ITT – Full accounting of subjects: Yes (no drop-outs) • Primary and additional analyses – Predefined analyses presented • Appropriate statistical comparisons • Magnitudes and precision of effect – 95% CIs of difference for all; p-value for primary outcome • Subgroup analyses (choice of subgroups justified) – Reasons given; ORs of subgroups compared (interaction test >0. 05) – Multivariable analysis post hoc due to somewhat unequal distribution of potential confounders
CONCLUSIONS • Do results support conclusions • Limitations discussed – Potential sources of bias – Magnitude and precision of results – Generalizability • Results placed in context of current knowledge and practice – Consider outcomes not studied • e. g. , cost, availability, ease of use, other risks – Consider other relevant evidence outside study
CONCLUSIONS • Do results support conclusions: yes • Limitations discussed – Potential sources of bias: probably not – Magnitude and precision of results: Not precision – Generalizability: Yes • Results placed in context of current knowledge and practice – Consider outcomes not studied • No (safety, time to d/c, willingness to repeat studied) – Other relevant evidence outside study • Prior study; information on the drugs