Phylogenetic comparative methods Comparative studies nuisance Evolutionary studies
 Evolutionary studies (objective) Community ecology (lack of alternatives)")



IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods,")
variances that are known Phylogeny")























 OU process �measures the strength of signal")


 OU process �measures the strength of signal")
IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods,")






")
IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods,")














- Slides: 57
Phylogenetic comparative methods Comparative studies (nuisance) Evolutionary studies (objective) Community ecology (lack of alternatives)
Current growth of phylogenetic comparative methods New statistical methods Availability of phylogenies Culture
One of many possible types of problems or as a special case This model structure can be used for a variety of types of problems
Assumptions: y takes continuous values x can be a random variable or a set of known values (continuous or not) y is linearly related to x are random variables with expectation 0 and finite (co)variances that are known
Statistical methods (P)IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods, not models) Other methods for other statistical models ML, REML, EGLS, GLMM, GEE, “Bayesian” methods
e are random variables with expectation 0 and finite (co)variances that are known Phylogeny provides a hypothesis for these covariances
Close Relatives Tend to Resemble Each Other
What does this represent? How is it constructed? Is it known for certain?
Assume that this represents time and is known without error Translate into the pattern of covariances in among species V
Trait value Hypothetical trait for a single species under Brownian motion evolution possible course of evolution Time
Trait value another possible course of evolution Time
Trait value another possible course of evolution Time
Trait value Brownian motion evolution gives the hypothetical variance of a trait Variance Time
Trait value Brownian motion evolution Variance Time
Brownian motion evolution of a hypothetical trait during speciation
Variance between species = Time
Total variance = Total time Variance between species = Time
Total variance = Total time Covariance = Shared time Variance between species = Time
Brownian motion Covariance matrix giving phylogenetic covariances among species diagonal elements give the total variance for species i off-diagonal elements give covariances between species i and species j
1. I am confused by the authors use of "branch lengths" on page 3023. I'm not sure if "different types of branch lengths" mean different phylogenetic analyses or something else I'm not aware of. 2. Digression - non-Brownian models of evolution
Ornstein-Uhlenbeck evolution Stabilizing selection with strength given by d
Variance between species < Time
Total variance << Total time Variance between species < Time
Ornstein-Uhlenbeck evolution Time Variance Stabilizing selection means information is “lost” through time Phylogenetic correlations between species decrease
Phylogenetic Signal (Blomberg, Garland, and Ives 2003) OU process �measures the strength of signal
Assumptions: y takes continuous values x can be a random variable or a set of known numbers y is linearly related to x e are random variables with expectation 0 and finite (co)variances that are known If d must be estimated, cannot be analyzed using PIC or GLS
1. If we are dealing with a recent, rapid radiation, (supported clade but with short branches) will the lack of branch length data render any PIC not very informative biologically, because we would expect non-significant probabilities, based solely on the branch lengths alone? page 3022, second paragraph.
Phylogenetic Signal (Blomberg, Garland, and Ives 2003) OU process �measures the strength of signal
Statistical methods (P)IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods, not models) Other methods for other statistical models ML, REML, EGLS, GLMM, GEE, “Bayesian” methods
PIC 4 1 y 4 2 3 y 1 y 2 y 3
4 1 y 4 2 3 y 1 y 2 y 3
PIC Regression through the origin
PIC You could also use different branch lengths for x:
Branch lengths of y Branch lengths of x
PIC You could also use different branch lengths for x: When could this be justified?
When could this be justified? Never (? )
Statistical methods (P)IC = GLS Phylogenetic independent contrasts Generalized Least Squares (these are methods, not models) Other methods for other statistical models ML, REML, EGLS, GLMM, GEE, “Bayesian” methods
Elements of V are given by shared branch lengths under the assumption of “Brownian motion” evolution
Generalized Least Squares, GLS
Ordinary least squares V=I
Related to ordinary least squares
Values of are linear combinations of yi
1. If IC and GLS can yield identical results and the authors refer to IC as "a special case of GLS models" (p. 3032), in what situation(s) would GLS be a more appropriate method? In other words, why not just use IC?
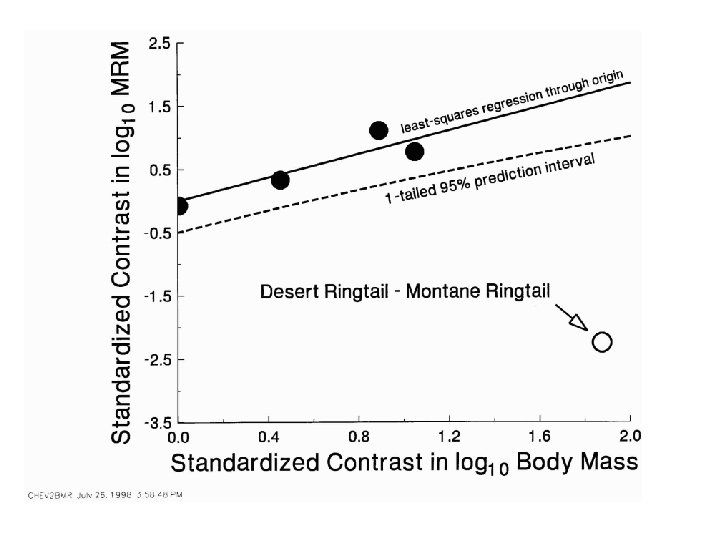
Divergence time for desert and montane ringtail populations assumed to be 10, 000 years
Predicting values for ancestral and new species
Is the prediction of the estimate of y for species I more or less precise than what you would expect from a standard regression analysis?
1. When dealing with multiple, incongruent gene trees, we can perform multiple PIC's on each tree, and find a correlation or not. How do we know which is the "right" answer? 2. The three main phylogenetically based statistical methods described in the reading (IC, GLS, and Monte Carlo simulations) rely on correct information about tree topology and branch lengths. If we are unsure of the correctness of these basic assumptions, what is the best way to analyze our data?
1. I'm unclear how data can be statistically significant when transformed, but not significant otherwise. This seems like cheating/lying. The paper discussed researchers' decisions about branch lengths, especially in terms of transformations (OU, ACDC). Do researchers use ultrametric trees for these analyses?