PHARMACOKINETICS PASSAGE OF DRUGS ACROSS CELL MEMBRANES How

PHARMACOKINETICS

PASSAGE OF DRUGS ACROSS CELL MEMBRANES

How do drugs cross cell membrane ? A. Passive methods of transport: cell is passive ; it has no active role and no energy is needed. 1. Simple lipid diffusion: most important passive method. Free drug molecules pass down their concentration gradient from high concentration outside the cell to low concentration inside cell across lipid bilayer of cell membrane. Drug molecules must be lipid soluble. Lipid solubility of drug molecules is measured by octanol/water partition coefficient; the higher it is, the more lipid soluble are drug molecules, & the more is their diffusion rate across the lipid cell membrane.

Fick”s Law of diffusion: this predicts the rate of movement of free molecules across a barrier membrane. The rate of diffusion is calculated from: concentration gradient of free drug molecules C 1 – C 2 , area of barrier membrane, its thickness, & the permeability coefficient of membrane for drug molecules. Rate of diffusion (No. of free molecules moving across cell membrane / unit time) = (C 1 – C 2). Permeability coefficient. Area Thickness. MW Diffusion rate follows first order kinetics. Molecules that are large and water soluble can not or poorly cross cell membranes by simple diffusion. Thus drug absorption into blood by simple diffusion is faster from intestine (large surface area) than from stomach (smaller surface area), and from lung alveoli (thin alveolar membrane) than from skin (thick barrier).

Most drugs are weak electrolytes, they dissociate poorly and this is affected by p. H of the medium. Henderson-Hasselbach equation estimates the [H+] or p. H of solutions of weak organic acidic or basic drugs : [H+]= K. (protonated form of molecule/deprotonated form) p. H= p. Ka – log (protonated molecule / deprotonated form) p. Ka – p. H = log (protonated / deprotonated) for weak organic acids or bases p. Ka is the dissociation constant of drug; it is equal to the p. H at which 50% of drug is dissociated. UNCHARGED drug MOLECULES ARE LIPID SOLUBLE (lipophillic; non-polar or less polar), AND THUS EASILY DIFFUSIBLE.

For acidic drugs For basic drugs p. Ka - p. H = log ( [HA] / [A-] p. Ka - p. H = log ( [BH+] / [B] HA acid p. H in stomach H+ + A- BH+ B + H+ alkaline p. H in intestine

For weak organic acids: e. g. aspirin p. Ka= 3 At gastric p. H of 1 , 3 -1 = log (HA / A-) 2 = log (HA/A-) 100 = (HA/A-) = 100/1 i. e. HA is about 100 times more than AAt gastric p. H of 2, HA is about 10 times more than AAnd vise versa at alkaline p. H For weak organic bases: e. g. ephedrine p. Ka = 6 ; At intestinal p. H of 8 , 6 -8 = log (BH+ / B) -2 = log (BH+/ B) 2 = log (B/BH+ ) On removing logs: 100 = (B / BH+) = 100/1 , so B is about 100 more than BH+ If p. H in intestine is 9 , then B is about 1000 times more than BH+. And vise versa at acid p. H.

2. Aqueous diffusion or filtration: across pores between endothelial cells in tissue capillaries or renal glomeruli. The driving force is the drug concentration gradient and capillary hydrostatic pressure being resisted by plasma colloidal oncotic pressure due to plasma albumin, and tissue pressure. Drug molecules with large MW or bound to plasma albumin are not filtered 3. Fascilitated diffusion : Molecules of some drugs may have certain structural configuration that allows facilitated diffusion e. g. glucose or other sugars. This uses a specific transmembrane carrier protein. No energy is needed because movement of molecules is not against the concentration gradient e. g. release of accumulated glucose into extracellular space adjacent to blood capillary. Facilitated diffusion is saturable & can be inhibited by cell injury.

B. Specialized methods of drug transport: Cell plays an active role. 1. Active transport : energy ± transmembrane carrier protein are provided by cell. The energy used for primary active transport comes from ATP breakdown via ATPase. Active transport of drugs is characterized by : - occurs against concentration gradient - Saturable ( limited capacity) - Can transport water soluble substances - Competitive inhibition may occur between drugs e. g. inhibition of active secretion of penicillin into urine by probenecid in cells of proximal renal tubules. - non-competitive inhibiton may occur by cell injury

Examples of active transport are : active transport of amino acids, like phenylalanine or L-DOPA and the drug alpha-methyldopa in intestine for absorption; expulsion of sodium ions from cells by sodium pump. 2. Pinocytosis : a. Exocytosis : it requires calcium; e. g. release of neurotransmitters e. g acetylcholine or noradrenaline by nerve impulses from vesicles inside the nerve endings or histamine release from granules inside mast cells during acute Ig. E-mediated allergic reactions. b. Endocytosis: is used for larger molecules that can not be transported by active transport or facilitated diffusion.

It permits molecules of proteins or hormones like insulin or complexes of smaller water soluble molecules (e. g. iron, vitamin B 12) with their specific transport proteins (e. g. transferrin or gastric intrinsic factor, respectively) to enter cells. The substance binds to specific receptor on cell membrane followed by infolding of that area of membrane around the substance and its subsequent internalization as vesicle inside the cell; contents are then released into cytosol after lysis of vesicle membrane. Strongly ionized substances e. g. quaternary amines are poorly transported by endocytosis

ADMINISTRATION AND ABSORPTION OF DRUGS

Drug administration: Route by which a drug is given. Drug absorption: This is the process by which drug crosses cell membranes from site of administration to reach local tissues for local action or to reach capillary blood and then systemic blood for systemic action in body. Administration for local effect: drug spreads in local tissues for local action; there is little systemic absorption, so little systemic side effects. Topical application Local injection inhalation Administration for systemic effect: Enteral administration (via GIT) : Oral by swallowing Sublingual Parenteral administration : Injection Inhalation Rectal Percutaneous

Administration for local effect : Dose forms or pharmaceutical formulation: depends on site of application: Topical application : Skin: lotions, sprays , powder, gel cream, ointment. Mucous membranes: Mouth and throat: gargles, lozenges, paints, sprays Eye, ear, nose: drops, ointments.
 Bronchi: inhalation of")
Colon: enema. Anorectal: ointments, suppositories Vagina: creams, tablets, pessaries (vagina suppository) Bronchi: inhalation of aerosol by either : Pressurized metered-dose inhaler PMDI: dry aerosol OR Nebulizer (needs compressor): wet aeresol. Local injection: aqueous or occasionally oily solution or sometimes drug suspension are used. Intra-thecal injection(into CSF); Intra-articular (into synovial cavity of joints); Intra-cardiac injection; intra-lesional; intra-pleural injection; intraperitoneal.
 : A.")
Administration of drugs for systemic effect : 1. Enteral administration (via GIT) : A. Oral ingestion by swallowing: dose forms or drug formulations are either liquids, tablets, or capsules. Absorption occurs mainly in upper small intestine, usually by simple diffusion. Weak Basic drugs: absorbed easily in small intestine since its molecules are largely uncharged at alkaline p. H, thus more lipid soluble and easily absorbable. Weak Acidic drugs : are partly absorbed in stomach since drug molecules are highly lipid soluble at acid p. H due to poor dissociation; a large portion may be absorbed in small intestine due to large surface area for absorption

Highly Water soluble drugs e. g. aminoglycoside antibiotics are poorly absorbed. e. g. streptomycin Quaternary ammonium compounds are always charged due to their quaternary nitrogen, so are poorly absorbed. (Note: Primary, secondary, and tertiary amines are lipid soluble in alkaline p. H of small intestine, thus easily absorbable by diffusion) Some drugs are absorbed by active transport e. g. vitamin D, L-DOPA. Systemic effect is delayed for about 20 minutes after drug swallowing due to GIT transit, and absorption of drug molecules in intestine followed by their passage via portal vein to liver before finally reaching systemic blood via hepatic veins.
. What is")
Drug X is a weak acid (p. Ka = 4. 4 ). What is the total concentration of drug X in plasma (p. H = 7. 4) if the concentration of its uncharged form HA is 1 micromol? In plasma : 4. 4 – 7. 4 = - 3 = log [HA] / [A-] , 3 = log [A-] / [HA]; On removing logs: 1000 = [A-] / [HA] [A-] plasma = 1000 HA = 1000 x 1= 1000 micromol Total concentration= [HA]+[A-] =1 +1000 = 1001 micromol

Factors affecting absorption from GIT: 1. Related to drug : A. Pharmaceutical factors : Dose form: Liquid or syrup is quickly absorbed Tablets must undergo fragmentation , desintegration, & dissolution into liquid before drug can be absorbed. A tablet is a mixture of active substance or drug with excipients, usually in powder form, & is packed under pressure to rounded or oval tablets or other geometric forms of various sizes. Excipients include : Fillers: e. g. inert substance e. g. lactose or starch that provide enough bulk to make handling and swallowing of tablet easy

Lubricants: as flow aid Desintegrants: Ensure tablet breakdown in GIT fluids e. g. starch, sodium bicarbonate Sweeteners: To mask bad taste or give a sweet taste Colouring agent: pigment or dye to give visual attraction. A coating material may be added to make tablet resistant to environment and avoid gastric irritation. Note: Effervescent tablets are dissolved in water before ingestion as liquid, and are thus considered to belong to the liquid preparations

Capsules: the most efficient method to take drugs orally The ingredients are enclosed in relatively stable shell : Hard-shell : used for dry powder ingredients or granules Soft-shell : for oils or active ingredients suspended in oil Both shells are made from aqueous solution of gelling agent such as : 1. Animal skin protein (collagen): mainly gelatin 2. Plant polysacharide derivative: like cellulose, carageenan Other ingredients added to gelling agent solution are : Plasticizers: e. g. glycerine or sorbitol to decrease hardness of shell Preservatives, Colouring agents and Surface treatment
-release preparations lead to slow")
Modified release preparations: include: 1. Slow-release or extended (or sustained)-release preparations lead to slow rate of drug absorption, so lower maximum concentration in plasma, but longer duration of action. 2. Controlled release preparation release drug at fixed rate for absorption to get more predictable plasma level. 3. Delayed release means drug is not available for absorption in upper small intestine after administration, but is released to be absorbed lower down later e. g. in colon 2. Enteric-coated tablets or capsules : The enteric coating, usually gelatin, dissolves slowly in intestine and thus release drug for absorption. Has longest lag time for absorption. It avoids gastric irritation & destruction of drug by gastric acid or pepsin in stomach. Avoid formation of non-absorbable drug-food complexes. Can hide unpleasant taste of drug, & extends its shelf life.

Particle size : e. g. absorption particle size. Pressure in making tablet: if high, tablet desintegration is decreased by large pressure B. Other factors: Chemical nature of drug (weak acidic or basic). neutral drug lacking ionizable group like digoxin & steroids Lipid solubility: affected by extent of ionization in solution and p. H of medium; lipid soluble nature of drug due to its organic structure; alcohol. 2. Related to GIT : Food intake; Food may reduce drug absorption from intestine due to dilution effect or binding to food proteins; fat in food delays gastric emptying; calcium in milk bind to & reduce absorption of tetracyclines

Gastric acidity and proteolytic enzymes may destroy & inactivate drug e. g. penicillin G, heparin, peptide hormone Gastric emptying: enhanced by glass of water, also by the drug metoclopramide which enhances gastric transit across the pylorus, and thus enhances intestinal absorption; but gastric emptying is delayed by exercise or by antimuscarinic drugs (which would reduce rate of absorption by intestine) or during pregnancy due to hormonal action. Intestinal motility: enhanced by diarrhea or osmotic laxatives e, g. magnesium sulfate which allows less time for absorption. Malabsorption: due to disease of intestine, pancreas, or biliary obstruction. Intestinal blood flow: favours quick absorption Drug interaction: e. g. antacids reduce iron absorption

After absorption, drugs pass from intestinal wall to portal venous blood then to liver before reaching hepatic veins to go to systemic blood and becomes bioavailable to tissues. Thus drugs may be exposed to first pass or pre-systemic metabolism or elimination in : a. Wall of small intestine: few drugs b. Liver: This is main site for most drugs, and is thus called first-pass hepatic metabolism or elimination (FPHE). Implications of FPHE: 1. Large FPHE of some drugs (e. g. propranolol, morphine, lidocaine) reduces the amount of these drug that reaches the systemic blood after its absorption i. e. it reduces their oral fractional systemic bio-availability (oral F). While drugs with low FPHE e. g. warfarin, theophylline, phenytoin, would have a higher oral F

2. With large FPHE, much larger therapeutic dose of a drug is needed orally than by parenteral injection. 3. In liver diseases, FPHE will be decreased and thus oral F will be increased; this demands reduction in oral dose of drug with large FPHE to avoid rise in its plasma level. 4. Hepatic microsomal enzyme inducers increase FPHE, and so decrease oral F of drugs; this needs an increase in oral dose of a drug with large FPHE to avoid decrease in its plasma level which would decrease its therapeutic effect, while hepatic microsomal enzyme inhibitors have the opposite effect.

The magnitude of FPHE may be measured by liver extraction ratio of drug ( ER ) : ER = (Cin – Cout ) / Cin where Cin is drug concentration in portal vein blood entering liver after absorption from intestine, and Cout is drug concentration in hepatic vein blood leaving liver. The effect of ER on oral F of drug may be estimated by: F = (f – f. ER) or F = f(1 – ER) where f is the fraction of oral dose absorbed from gut into portal blood. If 100% of drug is absorbed , then f = 1, so F = 1 – ER. If 80% of drug is absorbed, then F= 0. 8(1–ER) Liver Clearance of drug in ml/min or L/h = Q x ER where Q is hepatic blood flow rate/min either in ml/min which is about 1300 ml/min or in L/h.

Drug X was given oral by swallowing in a dose of 600 mg. If its fractional absorption from intestine is 0. 5 & ER is 0. 6 , calculate its F & Plasma concentration? Suppose plasma volume is 3000 ml. F = f(1 -ER) = 0. 5 (1 -0. 6) = 0. 2 or 20% Amount of dose that is absorbed to systemic blood plasma = Dose. F = 600 x 0. 2= 120 mg Plasma concentration = 120 / 3000 = 0. 04 mg/ml = 0. 04 x 1000 = 40 microgram /ml
 of an oral")
Oral systemic bioavailability: It is the fraction F or percentage (%) of an oral dose of drug that reaches the systemic blood after its oral administration. Fractional oral systemic bioavailability (F) = AUC oral dose / AUC i. v. of same dose. Percent F (%) = F. 100 AUC is area under concentration-time (C-T) curve. It represents amount of drug in plasma. F measures the extent of systemic bioavailability. Usually F is < 1 after oral drug administration by swallowing Factors affecting oral systemic bioavailability of drugs: Pharmaceutical factors GIT factors affecting absorption FPHE & factors affecting it

It is important to distinguish the rate of availability from the extent of availability. The rate of absorption and thus rate of availability is measured by the time needed to get the peak or maximal concentration in plasma after its oral administration. When drug absorption is rapid, then the time needed to reach maximum plasma level is shorter when compared to the slow absorption of the same drug dose if given as slow-release preparations
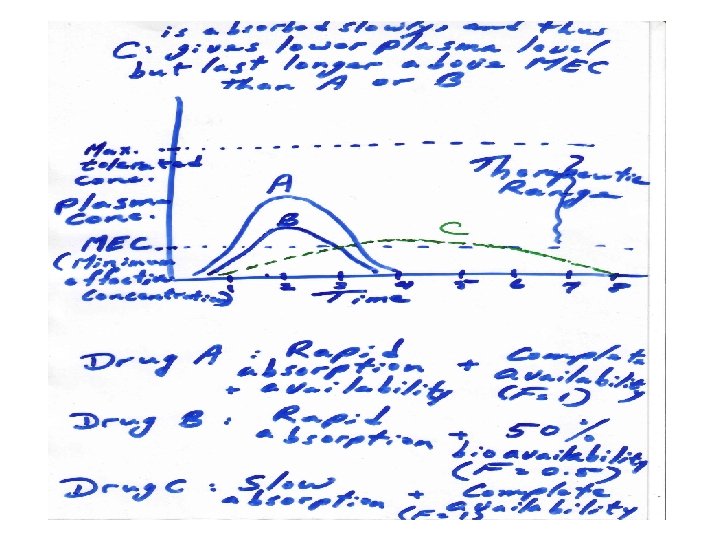

Bioequivalence : a pharmaceutical term used to denote that the extent and rate of bioavailability of one drug formulation are similar to that of another formulation of same drug. i. e. pharmaceutical bioequivalence. Such bioequivalent drug formulations are usually also therapeutically bioequivalent. It is needed when the drug is synthesized by a different manufacturer or when a different method of its synthesis have been employed.
 administration: Dose forms: tablets (usually), lingual spray occasionally - Quick effect")
B. Sublingual (SL) administration: Dose forms: tablets (usually), lingual spray occasionally - Quick effect (few minutes) due to direct absorption into systemic capillary blood under tongue. GIT and liver are by-passed, so FPHE is avoided. Systemic bioavailability is high. e. g. GTN Glyceryl trinitrate tablets SL for relief of acute angina pectoris. C. Rectal: Dose forms: suppositories (drug in wax or coconut oil) melt at core body temperature to release drug for absorption. Absorption from lower 1/3 of rectum directly to systemic blood, but from upper rectum it can go to liver by portal blood so exposed to FPHE. Absorption can be variable from lower rectum. Quick effect occurs. Indicated in: Vomiting. Children esp. with high fever. Gastric irritants esp. in PU. Coma.

2. Parenteral administration for systemic effect: A. Injection: Bioavailability is high. Dose forms either aqueous solution or sometimes drug suspension in vials or ampules. Sometime it comes as powder or crystals in vials to be reconstituted by suitable solvent before injection. Occasional comes as drug in oil. 1. Subcutaneous (SC): absorbed into systemic blood in capillaries of subcutaneous tissue. Effect occurs in 7 -10 min. Avoid irritant drugs and not suitable for large volumes. Sites: upper outer arm; Sometimes front of thigh or anterior abdominal wall. 2. Intramuscular (IM): effect comes in about 5 min. Sites: Upper outer gluteal region (gluteus maximus) ; Upper outer arm (deltoid muscle). Irritant drugs may be given IM (causes local pain), and IM route may be useful for large volumes.
: slowly done; immediate effect since no absorption. 100% bioavailability. Large volumes")
3. Intravenous (IV): slowly done; immediate effect since no absorption. 100% bioavailability. Large volumes may be given. Fast injection may be dangerous with some drugs due to hypotension. or cardiac arrhythmia that may be fatal. Sites: antecubital vein; avoid leak outside vein as some drugs are irritant e. g. aminophylline. Factors affecting systemic absorption after injection: Tissue blood flow: Absorption is retarded by cooling area or in shock (esp. with SC). In shock, avoid SC; usually IM or IV are used in shock. Absorption enhanced on heating area by rubbing, or hot bath or exercise. Dose form: absorption is quicker with aqueous solutions than with suspensions; slow absorption also occurs with oily preparation or pellets. Hyaluronidase: This enzyme is given locally. It depolymerizes hyaluronic acid in ground substance, thus enhancing drug spread into tissue for quicker absorption Binding of drug to tissue: few drugs e. g. diazepam IM which binds to muscle and this slows absorption, and thus leads to delayed effect.

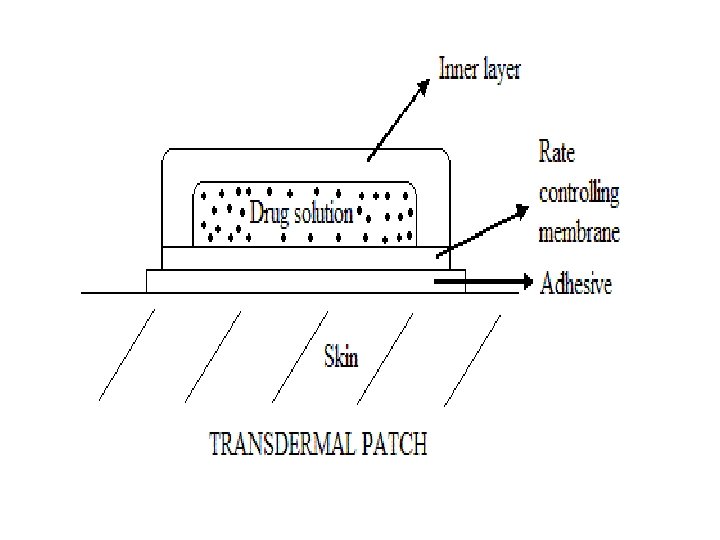
B. Inhalation : e. g. general inhalation anaesthetics are either vapours of volatile liquids or gases. Absorption depened on lipid solubility, blood solubility, alveolar ventilation, & pulmonary blood flow. Effect comes quick C. Skin: Stratum corneum with its keratin content is main resistance to drug absorption. Damaged skin e. g. burns can absorb any substance even if water soluble. Wet skin esp. if thin absorbs more than dry or thick skin. Percutaneous: ointment applied to thin skin e. g. to front of chest. Absorption is quick leading to quick effect e. g. GTN Transdermal: Synthetic transdermal patch containing drug is applied to skin; it slowly releases drug e. g. GTN patch for slow absorption to prolong its systemic effect.
- Slides: 37