PHARMACOCINETIQUE et MESURE de BIOEQUIVALENCE I PHARMACOCINETIQUE I

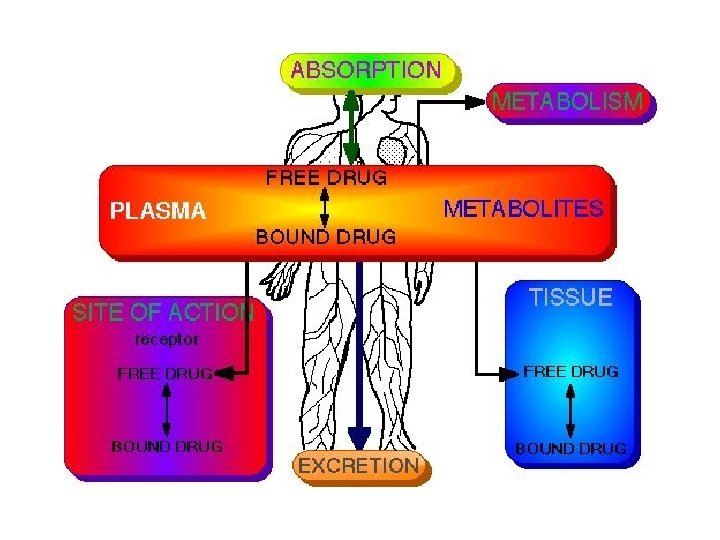
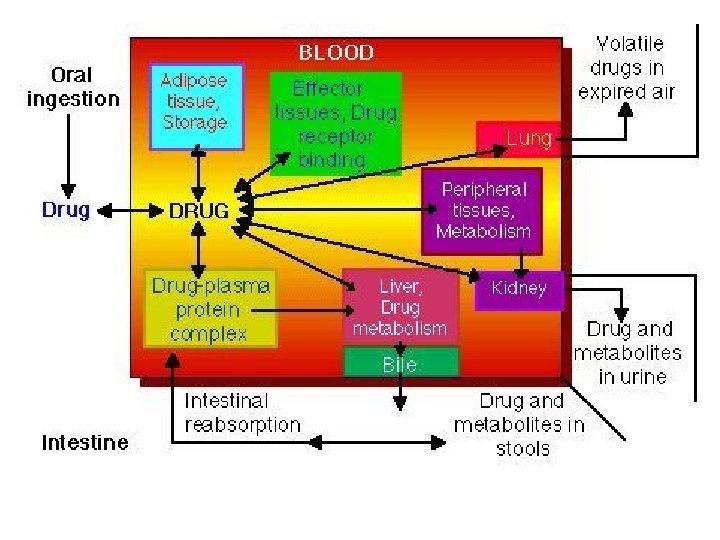
PHARMACOCINETIQUE et MESURE de BIOEQUIVALENCE I. PHARMACOCINETIQUE I. 1 - Définition I. 1. a - Définition de la pharmacocinétique La Pharmacocinétique a pour objet l'étude descriptive et quantitative du devenir d'un médicament dans l'organisme auquel il est administré. Alors que la Pharmacologie s'intéresse à l'action du médicament sur l'organisme, la pharmacocinétique concerne l'action de l'organisme sur le médicament. La pharmacocinétique clinique correspond à l'application des notions théoriques de pharmacocinétique au traitement des maladies par les médicaments. (Ajustement de posologie, . . . )
![pharmacocinétique pharmacodynamie Absorption Distribution [ ] effets Métabolisme Elimination 2](http://slidetodoc.com/presentation_image/1d72266eb64de2c1f69471f776ed942c/image-2.jpg "pharmacocinétique pharmacodynamie Absorption Distribution [ ] effets Métabolisme Elimination 2")
pharmacocinétique pharmacodynamie Absorption Distribution [ ] effets Métabolisme Elimination 2

I. 1. b - Définition des processus mis en jeu q. Processus d’ordre 1 Après son administration, le médicament subit un certain nombre de processus permettant : • sa résorption, • sa diffusion dans les tissus, • son excrétion (voie urinaire, voie biliaire, . . . ) d'une façon générale ces processus sont dit d'ordre 1. Variation de concentration d. C = - ke C 1 (1) dt Variation du temps Constante de proportionnalité appelée Constante de vitesse d’élimination La vitesse de variation de la concentration du médicament par unité de temps est égale au produit de la concentration du médicament par la constante de vitesse de 1 er ordre (ke). Le signe négatif indique cette concentration diminue dans le temps.

I. 1. b - Définition des processus mis en jeu q. Processus d’ordre 1 Après son administration, le médicament subit un certain nombre de processus permettant : • sa résorption, • sa diffusion dans les tissus, • son excrétion (voie urinaire, voie biliaire, . . . ) d'une façon générale ces processus sont dit d'ordre 1. Variation de concentration d. C = - ke C 1 (1) dt Variation du temps Constante de proportionnalité appelée Constante de vitesse d’élimination La vitesse de variation de la concentration du médicament par unité de temps est égale au produit de la concentration du médicament par la constante de vitesse de 1 er ordre (ke). Le signe négatif indique cette concentration diminue dans le temps.
 La vitesse d'élimination du composé")
Il résulte deux conséquences de ce processus : 1) La vitesse d'élimination du composé est proportionnelle à sa concentration : plus la concentration du médicament est élevée, plus la quantité de ce médicament éliminée par unité de temps est grande. 2) Le logarithme de la concentration varie linéairement avec le temps : Par intégration l'équation d. C = - ke C 1 devient : dt ln Ct = ln C 0 - ke t t = temps Ct = concentration du médicament à l'instant t C 0 = concentration du médicament à l'instant t = Ø ke = constante de vitesse de premier ordre
 est de la forme")
L'équation (ln Ct = ln C 0 - ke t) est de la forme : y = - a x + b (-ke correspond donc à [-a], pente de la droite) ln (concentrations) concentrations temps L'équation (ln Ct = ln C 0 - ke t) CØ = C initiale -a = -ke = pente temps peut également s'écrire : ln Ct = - ket C 0 Autrement dit : Ct = C 0 e -ket Cette équation exprime le phénomène exponentiel des processus d'ordre 1.

q. Processus d’ordre 0 Élimination d’une quantité constante de la quantité contenue dans l’organisme, par exemple 0, 15 g/L sont éliminés chaque heure (quelque soit la concentration) Q [mg] 10 Q = Vd. [C] élimination (ou [C]) 5 c(t) = c 0 – k. t 0 0 2 4 6 temps [h] 8 10 Situation plutôt rare, rencontrée lorsqu’un système d’élimination travaille en condition de saturation. Ex: éthanol, acide salicylique 9

I. 2. - Voie I. V. rapide unique : modèle à un et deux compartiments I. 2. 1 - Evolution des concentrations plasmatiques L'étude pharmacocinétique d'un médicament se fait généralement par l'étude de la variation des concentrations plasmatiques ou sanguines en fonction du temps. La décroissance des concentrations plasmatiques en fonction du temps reflète le devenir du médicament dans l'organisme. I. 2. 1. a - Modèle monocompartimental La décroissance des concentrations expérimentales au cours du temps est assimilable à une mono-exponentielle. Dans ce cas la décroissance des concentrations exprime uniquement la phase d'élimination du médicament de l'organisme. On décrit alors l'évolution des concentrations (c)en fonction du temps (t) par une mono exponentielle.

Quantité dans le sang Bolus temps

Quantité dans le sang Bolus temps
 concentrations Ct = C 0 e-ket CØ = C à t=0 -a")
ln (concentrations) concentrations Ct = C 0 e-ket CØ = C à t=0 -a = -ke = pente IV temps Ct C 0= concentration extrapolée à t=0 (-ke)=constante de vitesse d'élimination. ke (-ke) représente la fraction de la dose de médicament présent dans l’organisme au moment t éliminée par unité de temps. Elle s'exprime en unité de temps – 1: si -ke = 0, 1 h-1, 1/10ème de la dose de médicament présent au moment t s'élimine par heure.

La représentation graphique en coordonnées semi-logarithmiques permet de linéariser la courbe. Plus que par sa constante de vitesse d'élimination, ce processus monoexponentiel se caractérise par sa demi-vie (= t ½) qui est le temps nécessaire pour que la concentration diminue de moitié. La demi-vie peut-être facilement calculée à partir du graphe des concentrations expérimentales ou extrapolée de Ct = C 0 e-ket t ½ = 0, 693/ke d’où ke = 0, 693 / t 1/2 ln C C C / 2 t 1/2 - Un tel modèle est dit à un compartiment. C'est l'unique cas où la pente de la courbe de décroissance des concentrations plasmatiques représente la constante de vitesse d'élimination (ke).

I. 2. 1. a - Modèle bicompartimental Dans un certain nombre de cas, la transformation en coordonnées semi-log donne le résultat suivant: La courbe d’évolution [C]= f (t) est la somme de 2 exponentielles

Quantité dans le sang Extravasculaire Sang Distribution Équilibre temps

Quantité dans le sang Extravasculaire Sang distribution élimination temps

Ce phénomène est en fait lié au devenir du médicament dans l'organisme : -après injection I. V. , le médicament se distribue d'abord dans les tissus, tout en subissant déjà un processus d'élimination : à cela correspond un premier segment. -La distribution terminée, le second segment correspond à un processus d'élimination pure.

Dose concentration tissus Phase de distribution et d’élimination sang Phase d’élimination temps

C t = A e - t + B e - t D IV Distribution (phase a) et élimination (phase b) Compartiment 1 Ct élimination (phase b) ke Compartiment 2 L’équation de la courbe est alors de type bi-exponentielle et où les coefficients et sont les pentes des phases de distribution et d'élimination. Il leur correspond une demi-vie de distribution et une demi-vie d'élimination : t ½ ( ) =0, 693 / et t ½ ( )=0, 693 /

On notera: 1 : contrairement à ce que l'on a pu voir pour le modèle mono, la constante , pente de la partie terminale, n'est pas la constante de vitesse d'élimination du médicament. Celle-ci est fonction des coefficients A et B et des exposants et : ke = (A + B) A + B 2 : une équation bi-exponentielle ne permet pas toujours de décrire la courbe de façon satisfaisante. Il est alors fait appel à des équations tri et d'une façon générale pluri-exponentielle.

Modèles à 3 compartiments 1 - plasma 2 - organes très vascularisés (cœur, foie, rein, cerveau) équilibre rapide 3 - organes peu vascularisés (graisse, tendons, cartilage) équilibre lent

graisse cerveau Sang Quantité dans le sang Modèle 3 compartiments distribution redistribution élimination temps

On notera que le nombre d'exponentielles décrivant au mieux la courbe expérimentale peut dépendre : - de l'intervalle de temps entre chaque prélèvement. - de la durée pendant laquelle les prélèvements sont effectués.
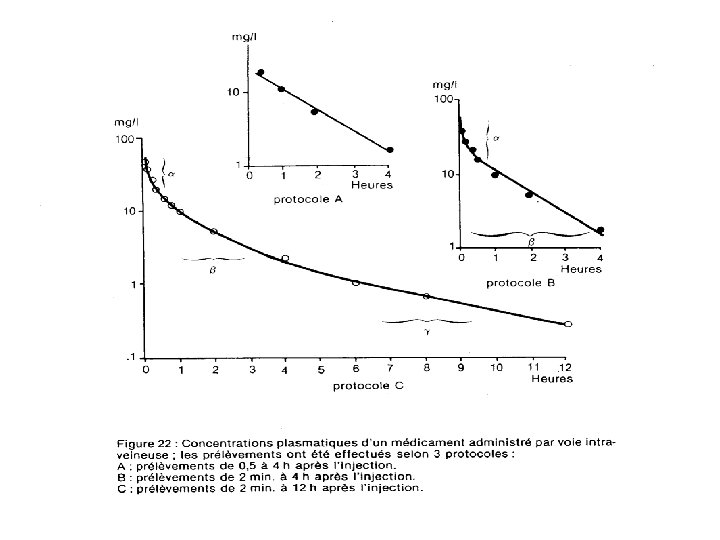

I. 2. 2 - Calcul des principaux paramètres pharmacocinétiques à partir des données plasmatiques I. 2. 2. 1. - Temps de demi-vie (t ½) Le plus souvent le terme de demi-vie sous-entend demi-vie d'élimination. Il ne faut cependant pas oublier qu'il existe autant de demi-vie que de phases identifiées sur la courbe expérimentale. La demi-vie est le temps nécessaire pour que la concentration plasmatique diminue de moitié ou encore le temps pour que la moitié du médicament soit éliminée de l'organisme. La moitié du médicament est éliminée en une demi-vie, il faut 5 t½ pour que 97 % de celui-ci soit éliminé de l'organisme. Après 7 t½ ils reste moins de 1% du médicament dans l’organisme.

La demi-vie se calculera graphiquement à partir de la courbe des concentrations plasmatiques ou mathématiquement à partir de la pente terminale d'élimination. La t 1/2 s'exprime en min. , heures….

Si la demi-vie est très employée en pharmacocinétique car facile à calculer et "parlant", il ne faut pas oublier que c'est cependant un paramètre secondaire, car la demi-vie ne reflète pas seulement l'élimination du médicament mais aussi sa distribution. On montre en effet que la formule générale de la demi-vie est : t½ = 0, 693 x Vd = 0, 693 Clt ke On peut donc imaginer pour deux médicaments une t½ identique avec des volumes de distribution et des clairances totales (paramètres d'élimination) différents. De même pour un même médicament, dans des conditions physiologiques différentes, le fait que la t½ ne change pas n'implique pas la même chose pour le Vd et la Clt

Clairance totale

La demi-vie plasmatique est indépendante de la voie d’ administration
 C'est la")
I. 2. 2. 2 - Aire sous la courbe (SSC ; AUC) C'est la surface délimitée par les axes du graphe et la courbe des concentrations. l'AUC peut-être calculée sur un intervalle de temps déterminé ou extrapolé à l'infini. L'AUC correspond à la quantité de médicament qui est passée dans le sang. Deux méthodes de calcul sont possibles : A) La plus simple est la méthode graphique dite des trapèzes qui consiste à découper la courbe expérimentale en autant de trapèzes qu'il existe de points expérimentaux : Surface du trapèze = (C 1 + C 2) x (t 2 - t 1) 2 L'addition des trapèzes permet d'obtenir la SSC des points expérimentaux.
 Concentration plasma (mg/l)")
Temps (heures) Concentration plasma (mg/l)
 C'est la")
I. 2. 2. 2 - Aire sous la courbe (SSC ; AUC) C'est la surface délimitée par les axes du graphe et la courbe des concentrations. l'AUC peut-être calculée sur un intervalle de temps déterminé ou extrapolé à l'infini. L'AUC correspond à la quantité circulante de médicament. Deux méthodes de calcul sont possibles : A) La plus simple est la méthode graphique dite des trapèzes qui consiste à découper la courbe expérimentale en autant de trapèzes qu'il existe de points expérimentaux : Surface du trapèze = (C 1 + C 2) x (t 2 - t 1) 2 L'addition des trapèzes permet d'obtenir la SSC des points expérimentaux.

Si la concentration au dernier temps de prélèvement n'est pas nulle, il est nécessaire d'extrapoler la surface entre le dernier prélèvement et l'infini selon la formule : AUC totale 0 --> = Ct / pente Les unités de surface sont le produit des unités de concentrations et de temps ex : µg x h x ml -1, mg x h x L-1. Trapèze 2 Trapèze 1 Trapèze 4 Trapèze 3 Ct
 On peut également calculer mathématiquement la surface sous la courbe. Elle représente")
B) On peut également calculer mathématiquement la surface sous la courbe. Elle représente l'intégrale de 0 à l'infini de l'équation exponentielle décrivant l'évolution des concentrations plasmatiques. Dans le cas d'un modèle à deux compartiments : Ct = A e - t + B e - t AUCo = (A e - t + B e - t) dt o AUCo = A/ + B/ N. B. : Le calcul de l'aire sous la courbe par la méthode des trapèzes ne fait pas intervenir le modèle mathématique proposé pour la courbe, sauf en ce qui concerne l'extrapolation à l'infini, on parle alors de modèle indépendant ou non compartimental, ceci est d'autant plus vrai que les prélèvements sont réalisés sur un temps suffisamment long, ce qui rend l'extrapolation à l'infini négligeable.
 Le volume apparent de distribution")
I. 2. 2. 3 - Volume de distribution (Vd) Le volume apparent de distribution représente un volume hypothétique dans lequel le médicament est uniformément réparti à la même concentration que celle mesurée dans le plasma.

Dose 10 mg Volume réel 500 ml Sans charbon Concentration 20 mg/l Volume apparent 500 ml Avec charbon actif Concentration 2 mg/l Volume apparent 5000 ml
 Le volume apparent de distribution")
I. 2. 2. 3 - Volume de distribution (Vd) Le volume apparent de distribution représente un volume hypothétique dans lequel le médicament est uniformément réparti à la même concentration que celle mesurée dans le plasma. C'est aussi le facteur de proportionnalité entre la quantité de médicament présent dans l'organisme et la concentration au même moment Qt = Ct x Vd => Vd = Qt/Ct Une façon simple de calculer le volume de distribution est de le calculer au temps t=0. Par I. V. la quantité Q 0 est égale à la dose injectée et dans ces conditions : Vd = Q 0 = Div C 0 où C 0 représente la concentration extrapolée au temps 0. Cependant cette formule n'est applicable que dans le cas particulier où l'administration du médicament a été réalisée par voie I. V. et où la décroissance des concentrations est mono-exponentielle.

Vd grand concentration tissulaires élévée/plasma distribution non homogène Vd petit Au minimum volume plasmatique = 0, 04 l/kg
 éthanol (petites")
Volume du Compartiment Exemple de molécules Eau eau totale (0, 6 l/kg) éthanol (petites molécules) eau extracellulaire (0, 2 l/kg) mannitol sang (0, 08 l/kg) plasma (0, 04 l/kg) héparine Graisses (0, 2 -0, 35 l/kg) thiopental (1, 5 -3 l/kg) Os (0, 07 l/kg) fluor, plomb

Dans le cadre d'un système bicompartimental, on peut calculer: - un volume central de distribution : V 1 = Dose C 0 A + B - volume de distribution extrapolé : Vd ext = Dose B - un volume de distribution appelé Vd ou Vdaire : Vd = Cl. t = F. dose AUC 0 . Le volume de distribution s'exprime en litres (ou ml) mais comme il peut être influencé par les caractéristiques de l'individu, on l'exprime aussi en L x kg-1.
 C'est le volume sanguin ou plasmatique")
I. 2. 2. 4 - Clairance plasmatique (Clt) C'est le volume sanguin ou plasmatique totalement débarrassé d'une substance par unité de temps. Elle se calcule selon l'équation suivante : Clt = F. dose en (ml min-1 ) ou (l h-1 ) éventuellement / kg. AUCo Mais aussi : Clt = ke x Vd en effet t 1/2 = 0, 693 x Vd donc Clt = 0, 693 x Vd Cl t 1/2 comme t 1/2= 0, 693/ke on a bien Cl= ke x Vd Cette formule est applicable quelque soit le mode d'administration du médicament si F est connue (F, fraction de la dose résorbée, est égale à 1 si injection intraveineuse. Voir le chapitre biodisponibilité). La clairance plasmatique totale correspond à la somme des différentes clairances en rapport avec les divers processus d'élimination. Clt = Cl hépatique + Cl rénale + Cl….

Cinétique de la vancomycine sujet normal

Cinétique de la vancomycine Insuffisant rénal

I. 3 - Voie orale : modèle mono-compartimental I-3 -1 - Evolution des concentrations plasmatiques Ct = -A e –kat + B e –ket t½ de résorption = 0, 693 et t½ d'élimination = 0, 693 ka ke Cmax 1 -3 -2 Calcul des principaux paramètres • Cmax et T max observés • C max et T max calculés T max = 1 x log ka - ke C max =FD e -ke Tmax Vd Tmax temps

ln. C Cmax C ka Vd élimination Q ke résorption + élimination Tmax Ct = -Ae –kat + Be –ket Lorsque t>>tmax alors Ct = Be –ket Temps

ln. C ka Distribution + élimination P C Vd B Q élimination ke Ct = -C 1 e-kat + C 2 e-at + C 3 e-bt Ct = -C 1 e-ka’t + C 2 e-ket avec ka’<<ka A Résorption + Distribution + élimination A : résorption rapide et décroissance monocompartimentale B : résorption très rapide. Les phases de distribution et d’élimination sont distinctes. Modèle bicompartimental.
 Elle")
I. 4 – Administration répétée I. 4. 1 - Perfusion veineuse (1 compartiment) Elle a pour but d'obtenir une concentration plasmatique stable. Elle se distingue de l'injection IV car l'apport en continu du médicament dans l'organisme conduit à une élévation constante des concentrations jusqu'à un certain niveau. C T K Vitesse de perfusion (constante) C VD ke Vitesse de d’élimination

Quantité dans le sang temps

Quantité dans le sang Perfusion continue Css 5 x T 1/2 temps Le niveau de Css est déterminé par la vitesse de perfusion

A - Plateau de concentration Durant la perfusion, deux processus sont en compétition : - la vitesse de perfusion (= entrée du médicament dans l'organisme). - la vitesse d'élimination (= sortie du médicament de l'organisme) La vitesse de perfusion étant constante, au niveau sanguin on note une augmentation de la concentration du médicament jusqu'à ce que la vitesse d'élimination égale la vitesse de perfusion : c'est l'état d'équilibre ou steady-state. Cet état ne bougera pas tant que la vitesse de perfusion restera constante et tant qu'aucun phénomène ne vienne perturber le processus d'élimination. - la vitesse de perfusion est constante (ordre zéro). - la vitesse d'élimination est d'ordre 1.

- la variation de la Q de médicament : d. Q = K - ke Q dt - par intégration : Q = K (1 - e - ke. T) ke où T = temps depuis le début de la perfusion exprimé en terme de concentration: C p = K (1 - e - ke. T) ke. Vd T e - ke. T 0 Cp ss = K = K ke. Vd Cl

- Le temps nécessaire pour atteindre le plateau ne dépend que de ke (ou t½ puisque t½ = 0, 693 / ke) Si t ½ est courte, le plateau sera rapidement obtenu et inversement. On montre qu'après 3, 3 t ½ on est à 90 % du plateau théorique et à 97 % après 5 t ½. - Le niveau de concentration du plateau ne dépend que de la vitesse de perfusion.

Temps nécessaire pour atteindre le plateau t t=t 1/2 t=2 t 1/2 t=3 t 1/2 t=4 t 1/2 t=5 t 1/2 t=6 t 1/2 t=7 t 1/2 t=8 t 1/2 t=9 t 1/2 fss 0, 5 0, 75 0, 87 0, 94 0, 97 0, 98 0, 996 0, 998 t=∞ 1, 000 Fss= fraction de steady state Fss=1 -(0, 5)t/t 1/2 En pratique plateau atteint après 5 t 1/2

Étude des concentrations plasmatiques après arrêt de la perfusion cpss Cpss cpt T N. B. : Dans les deux cas, la décroissance se fait de la même manière.

Après l’arrêt de la perfusion, l’ évolution des concentrations plasmatiques décrit une courbe de forme exponentielle. Ct ou Cpss

Étude des concentrations plasmatiques après arrêt de la perfusion cpss Cpss cpt C’p=cpss e-ke t’ C’p=cpt e-ke t’ T N. B. : Dans les deux cas, la décroissance se fait de la même manière.
 avant le plateau d'équilibre")
Étude des concentrations plasmatiques après arrêt de la perfusion a) avant le plateau d'équilibre Cp. T = K (1 - e - ke. T) ke. Vd puis diminution suivant un système mono-exponentiel : C’p = (Cp. T)e - ket' K (1 - e - ke. T) e -ke t' C'p = ke. Vd où t' = temps écoulé depuis la fin de la perfusion et C' concentration à t'.
 au plateau Cp = K = Cp ss ke. Vd C’p = (Cp")
b) au plateau Cp = K = Cp ss ke. Vd C’p = (Cp ss )e - ket' = K e - ket' ke. Vd où t' = temps écoulé depuis la fin de la perfusion.
 Clairance totale Cp ss = K = K Ke.")
Etude des paramètres pharmacocinétiques a) Clairance totale Cp ss = K = K Ke. Vd Cl Cl = _K_ Cp ss b) Demi-vie : correspond au temps pour obtenir la moitié de la concentration d'équilibre t ½ = 0, 693 ke c) Volume de distribution Vd = t ½ x Cl (équation générale) 0, 693

Dose de charge Quantité dans le sang D =Vd. Css Perfusion continue + Extravasculaire Sang Css Perfusion temps
 Dose de charge (D*) Dose de charge + perfusion Perfusion CPSS Dose de")
e) Dose de charge (D*) Dose de charge + perfusion Perfusion CPSS Dose de charge D* = Cpss x Vd Réaliser simultanément une perfusion de vitesse K=Cpss x Cl (-----) et une injection IV à la dose de charge D*=Cpss x Vd (…. . ) permet d’obtenir d’emblée et de maintenir la concentration souhaitée (____).

Quantité dans le sang Perfusion continue et modification de la vitesse Vp 5 x T 1/2 Vp/2 5 x T 1/2 temps

I. 4. 2 – Administration réitérée : Voie IV et Orale Les règles qui régissent l'évolution des concentrations du médicament lors de l'administration réitérée sont identiques qu'il s'agisse d'administration par voie I. V. rapide ou voie orale, au facteur F de biodisponibilité près. I. 4. 2. 1 - Évolution des concentrations plasmatiques L'intervalle de temps entre deux doses : et la dose est : D. Les concentrations obtenues après chaque dose s'additionnent. On observe au départ une augmentation des concentrations puis au bout d'un certain temps, la dose administrée compense la quantité de médicament éliminée dans l'intervalle .

On obtient un "pseudo plateau" ou les concentrations fluctuent entre : - une concentration minimale : Cmin obtenue juste avant une prise de médicament. - une concentration maximale : Cmax. Le temps nécessaire pour atteindre 97 % du plateau est de 5 t ½.

Concentrations plasmatiques Influence du rythme d’administration 3 mg une fois/jour 1 mg matin midi soir T 1/2=17 h J 1 J 2 J 3 J 4 J 5 J 6 Jours La diminution de fréquence augmente Cmax et diminue Cmin, ne modifie pas Cmoy Attention si marge thérapeutique étroite


Concentrations plasmatiques Influence de la demi vie T 1/2=17 h T 1/2= 8 h J 1 J 2 J 3 J 4 J 5 J 6 Jours Le délai pour atteindre le plateau et le niveau du plateau augmente avec la demi-vie
 autour")
I. 4. 2. 2 - Paramètres pharmacocinétiques La concentration moyenne à l'équilibre (Cmoy) autour de laquelle fluctuent les concentrations (analogue à la concentration au plateau durant une perfusion - Cpss -) est la concentration où le débit d'entrée (D/ ) est égal à la vitesse d'élimination. Cmoy = D x 1 pour perfusion. (Cpss = K/Clt) Clt Cmoy = FD x 1 pour V. O. Clt Les concentrations au plateau (Cmax, Cmoy, Cmin) sont : - proportionnelles à la dose D - inversement proportionnelles à l'intervalle entre les doses

On obtient un "pseudo plateau" ou les concentrations fluctuent entre : - une concentration minimale : Cmin obtenue juste avant une prise de médicament. - une concentration maximale : Cmax. Le temps nécessaire pour atteindre 97 % du plateau est de 5 t ½.
 entre Cmax et Cmin. FC = Cmax - Cmin Cmax")
Fluctuation des concentrations (FC) entre Cmax et Cmin. FC = Cmax - Cmin Cmax dépend des valeurs relatives entre et t ½. = t ½ < t ½ > t ½ FC = 50 % FC < 50 % FC > 50 %

Lorsque le t ½ du médicament est longue et qu'il est souhaitable d'avoir des concentrations rapidement efficaces, on peut réaliser une dose de charge (D*). D* = Vd x Cmoy } en fonction de la concentration F prise en référence ou D* = Vd x Cmax } F on peut alors calculer la dose d'entretien (D) C moy = FD x 1 donc Clt D entretien = Cmoy x Clt x x 1 F D* = Vd x Cmoy x F = Vd x t ½ D Cmoy x Clt x x F 0, 693 x Vd x D* = 1, 44 x t ½ = Rapport d’accumulation D Il est donc possible à partir de la dose d’entretien de calculer la dose de charge et inversement.

I. 5 – Biodisponibilité / Bioéquivalence I. 5. 1 - Définitions La biodisponibilité caractérise la forme pharmaceutique d'un médicament administrée chez un individu. Elle représente la manière dont le principe actif est mis à la disposition de l'organisme sans préjuger de son effet pharmacologique. Depuis 1972, la définition de la biodisponibilité est la suivante (OMS, FDA) : "La biodisponibilité représente la fraction du principe actif administré qui atteint la circulation générale et la vitesse avec laquelle l'atteint".

üIl y a donc un aspect quantitatif, c'est la fraction effectivement résorbée, c'est-à-dire le coefficient de résorption (F), évalué d'après l'aire sous la courbe (ASC, AUC) des temps/concentrations ou d'après la quantité de principe actif (U 0 - ) éliminé au niveau urinaire sous forme intacte… ü…et un aspect cinétique, représenté par la vitesse de résorption caractérisé par Ka (constante de vitesse de résorption) ou le Tmax, temps d'apparition du pic plasmatique.

• Ces paramètres qui caractérisent un médicament peuvent être variables avec les différentes formes galéniques d'un même médicament, mais aussi du fait de l'existence d'une variabilité intra et/ou inter-individuelle. • Deux médicaments présentant des critères de biodisponibilité identiques lorsqu'ils sont administrés à posologie égale à un échantillon d'individus sont dits bioéquivalents. La bioéquivalence garanti l'équivalence thérapeutique pour un malade (médicaments génériques).

Cependant des facteurs de variabilité liés aux patients peuvent modifier cette équivalence. La bioéquivalence est donc une condition nécessaire mais pas toujours suffisante à l'équivalence thérapeutique. I. 5. 2 – Biodisponibilité absolue et relative • La biodisponibilité d'un médicament administré par voie IV est totale et immédiate. • F, fraction du P. A. , disponible par l'organisme après administration par voie IV est donc égal à 1 par définition. • La biodisponibilité d'un médicament administré par : - voie différente que la voie intravasculaire peut être comparée soit à la voie IV, on parle alors de biodisponibilité absolue. - soit à une voie différente que la voie IV, on parle alors de biodisponibilité relative.

I. 5. 2. 1 - Biodisponibilité absolue Le calcul de la biodisponibilité absolue répond à la relation : AUCorale 0 - F = ---------- avec 0 < F < 1 AUCiv 0 - -Les deux administrations doivent être faites, si possible, à la même dose, aux même sujets de manière à éliminer les facteurs de variabilité interindividuels. Si pour des raisons de sécurité, on ne peut utiliser une dose iv aussi importante que par voie orale, il y a lieu de corriger la formule AUCorale 0 - Dose. IV F =----------- X -------- AUCIV 0 - Dose orale

- La biodisponibilité peut être de la même manière calculée à partir des données urinaires. U orale 0 - Dose IV F= ----------- X -------- U IV 0 - Dose orale

• La biodisponibilité absolue évalue l'intérêt d'une voie d'administration par rapport à la voie IV. • Elle correspond à un rendement de la résorption. • Elle fait partie des informations essentielles devant figurer dans le dossier d'A. M. M. d'un médicament lors de sa mise sur le marché, d'un changement de sa voie d'administration, d'un changement de posologie, de sa formulation.

I. 5. 2. 2 – Biodisponibilité relative Le calcul de la biodisponibilité relative d'un médicament répond à la relation : AUC forme B 0 - U 0 - forme B F = ------------ = --------AUC forme A 0 - U 0 - forme A ou : - B est la forme galénique à étudier. - A est la forme de référence qui doit être la forme présentant la meilleure biodisponibilité absolue, souvent la forme solution du médicament. - le choix de la forme de référence doit être adapté au but recherché : par exemple pour une nouvelle formulation, on prendra en référence la forme déjà commercialisée.

La biodisponibilité relative d'un médicament sera évaluée dans les cas suivants : • modification de formulation. • substitution d'une forme par une autre. • préparation d'un médicament générique. Dans ce cas, le médicament générique sera testé soit contre l'original, soit contre une solution du principe actif de biodisponibilité supérieure à l'original. Le but de cette évaluation de la biodisponibilité relative étant de montrer la bioéquivalence des deux formes. I. 5. 3 – Evaluation de la biodisponibilité Elle s'effectue par un essai comparatif randomisé in vivo, chez l'homme.
- Slides: 81