Pediatric ALL Abdulhadi Alzaben MD Pediatric hematology oncology

Pediatric ALL Abdulhadi Alzaben, MD Pediatric hematology oncology and stem cell transplant

INTRODUCTION • • • Acute leukemia, the most common form of cancer in children, comprises approximately 30 percent of all childhood malignancies. Acute lymphoblastic leukemia (ALL) being five times more common than acute myeloid leukemia (AML) Survival rates for ALL have improved dramatically since the 1980 s, with a current five-year overall survival rate estimated at greater than 85 percent.

DEFINITION Uncontrolled proliferation of immature blood cells with a different immunological subtypes which is lethal within 1 – 6 months without treatment. The disorder starts in the bone marrow, where normal blood cells are replaced by leukemic cells. Morphological (FAB), immunological, cytogenetic, biochemical, and molecular genetic factors characterize the subtypes with various response to treatment

EPIDEMIOLOGY • ALL incidence of approximately 3. 4 cases per 100, 000 • The peak incidence occurs between 2 -5 years of age. • Boys more than girls • Identical twins – If one twin develops leukemia during the first 5 years of life the risk of the second twin developing leukemia is 20%

EPIDEMIOLOGY Acute lymphoblastic leukemia: 75% Acute myeloid leukemia: 20% Chronic myeloid leukemia constitute 3% of all childhood leukemia and consist of: • Chronic myeloid leukemia ( CML) usually Philadelphia chromosome positive • Juvenile myelomonocytic leukemia (JMML) negative Philadelphia chromosome

ETIOLOGY The etiology of acute leukemia is unknown. The following factors are important in the pathogenesis of leukemia: • Ionizing radiation • Chemicals (e. g. , benzene in AML) • Drugs (e. g. , use of alkylating agents either alone or in combination with radiation therapy increases the risk of AML)

ETIOLOGY • • Increased incidence with the following genetically determined conditions: Shwachman–Diamond syndrome Ataxia telangiectasia Li–Fraumeni syndrome (germ line p 53 mutation) Neurofibromatosis Diamond–Blackfan anemia Kostmann disease Bloom syndrome
. 2. Fatigue (50%). 3. Pallor (40%).")
CLINICAL FEATURES General Systemic Effects: 1. Fever (60%). 2. Fatigue (50%). 3. Pallor (40%).
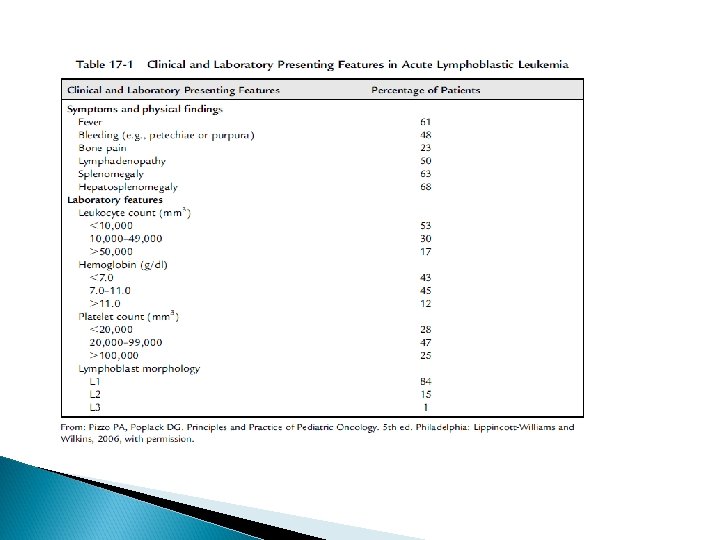

Hematologic Effects Arising from Bone Marrow Invasion Anemia – causing pallor, fatigability, tachycardia, dyspnea and sometimes congestive heart failure. Neutropenia – causing fever, ulceration of buccal mucosa and infection. Thrombocytopenia – causing petechiae, purpura, easy bruisability, bleeding from mucous membrane and sometimes internal bleeding (e. g. , intracranial hemorrhage).

Clinical Manifestations Arising from Lymphoid System Infiltration Lymphadenopathy – sometimes presents with bulky mediastinal lymphadenopathy causing superior vena cava syndrome Splenomegaly. Hepatomegaly.
,")
Clinical Manifestations of Extramedullary Invasion Central nervous system Although uncommon (<5 percent of cases), leukemia involving the central nervous system (CNS) can present with symptoms of increased intracranial pressure, including headache, vomiting, lethargy, and/or nuchal rigidity. Rarely, leukemia can present with cranial nerve abnormalities

Central nervous system Diagnosis by analysis of cerebrospinal fluid: CNS I: no lymphoblasts. CNS II: less than 5 cells/cm 3, but with leukemic blasts on centrifugation. CNS III: at least 5 cells/cm 3, with leukemic blasts on centrifugation or cranial nerve palsy

Mediastinum Enlargement due to leukemic infiltration by lymph nodes. May cause life-threatening superior vena cava syndrome (especially in T-cell ALL)

Testicular involvement Usually presents with painless enlargement of the testis. Occurs in 10– 23% of boys during the course of the disease at a median time of 13 months from diagnosis. Usually in T-cell ALL

Bone and joint involvement Bone pain initially present in 25% of patients Bone or joint pain, sometimes with swelling and tenderness due to leukemic infiltration of the periosteum.

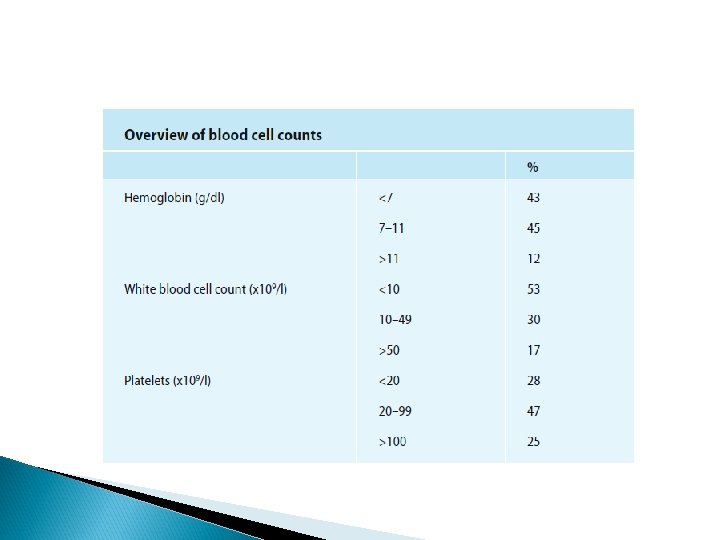
DIAGNOSIS Laboratory Studies Complete Blood count Hemoglobin: Moderate to marked reduction. White blood cell count: Low, normal, or increased Thrombocytopenia: 92% of patients have platelet counts below normal.

Blood smear: Blasts are present on blood smear. Very few to none (in patients with leukopenia). When the white blood cell (WBC) count is greater than 10, 000/mm 3, blasts are usually abundant.

Bone marrow: Bone marrow is usually replaced by 80 – 100% blasts. Megakaryocytes are usually absent. Leukemia must be suspected when the bone marrow contains more than 5% blasts.

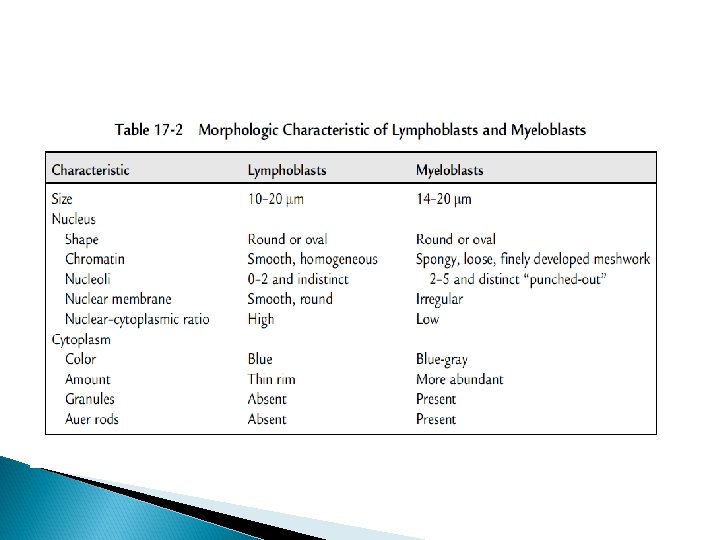
The hallmark of the diagnosis of acute leukemia is the blast cell, a relatively undifferentiated cell with diffusely distributed nuclear chromatin, one or more nucleoli and basophilic cytoplasm. Special bone marrow studies: Histochemistry Immunophenotyping Cytogenetics

Chest radiograph: Mediastinal mass in T-cell leukemia. Blood chemistry: Electrolytes, blood urea, uric acid, liver function tests. Cerebrospinal fluid: Chemistry and cells. Cerebrospinal fluid findings for the diagnosis of CNS leukemia
 and echocardiogram. Infectious disease profile: Varicella antibody titer, cytomegalovirus")
Cardiac function: Electrocardiogram (ECG) and echocardiogram. Infectious disease profile: Varicella antibody titer, cytomegalovirus (CMV) antibody titer, herpes simplex antibody, hepatitis antibody screening.

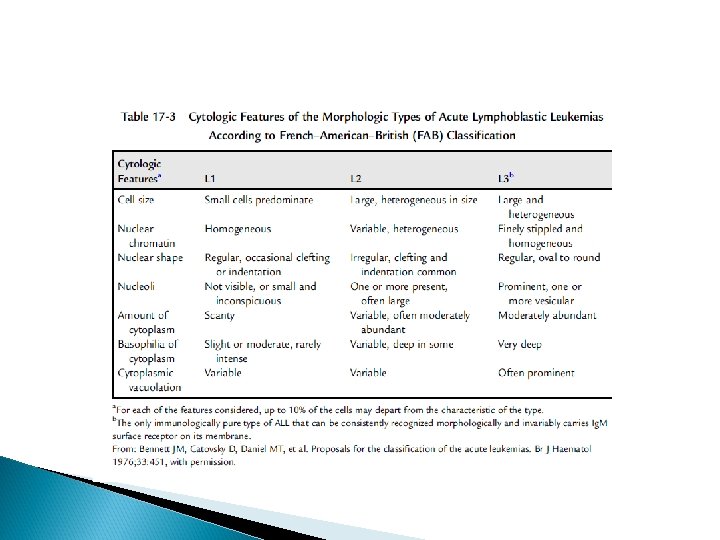
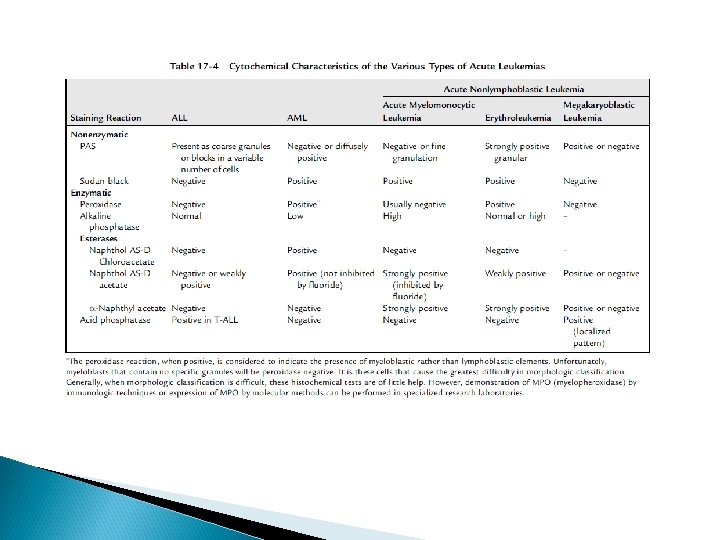
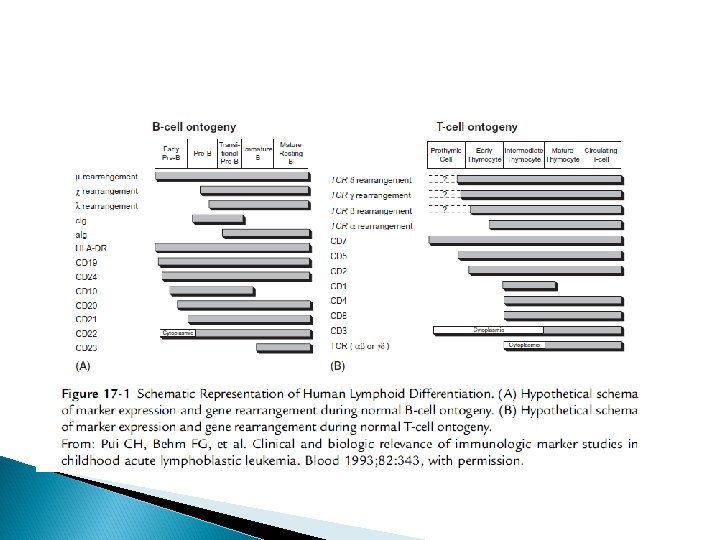
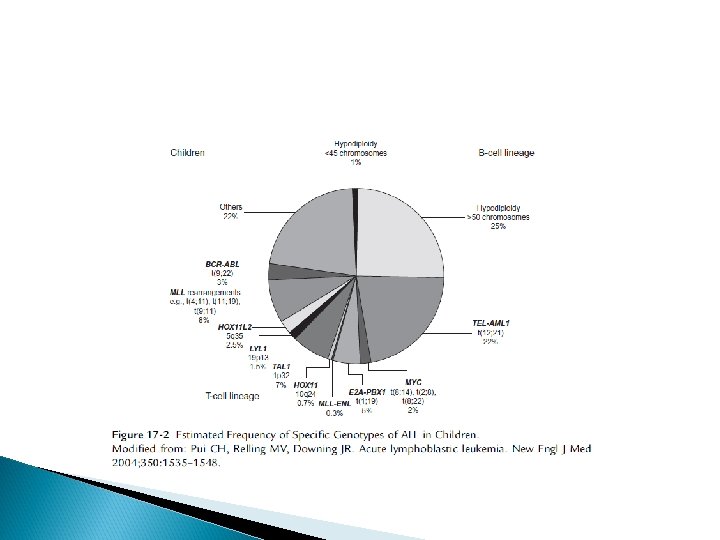
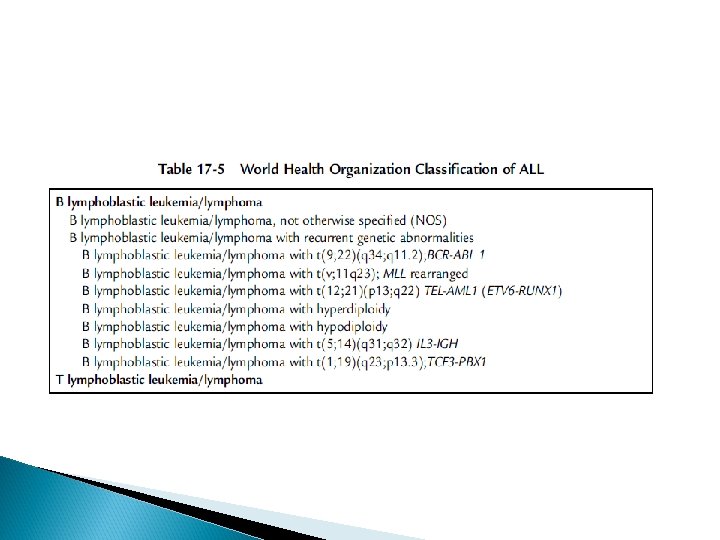
Classification Acute leukemia can be classified based on: morphologic characteristics cytochemical features, immunologic characteristics cytogenetic and molecular characteristics

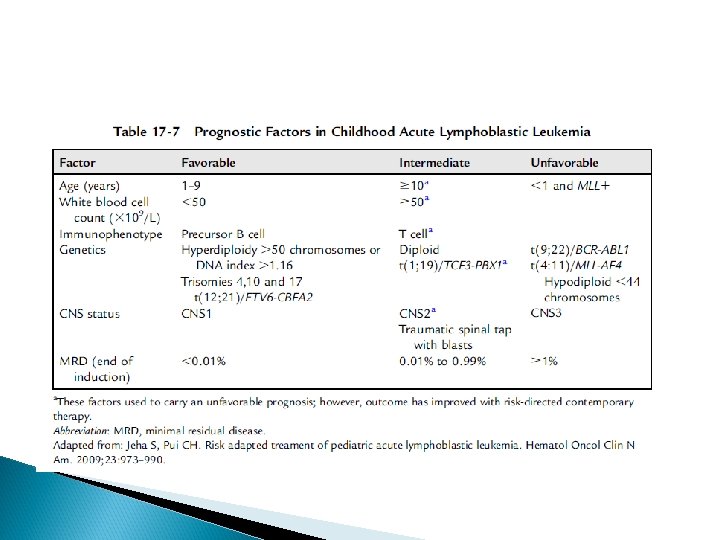
Clinical importance of immunological characterization: Eighty-five percent of children with common ALL (usually pre-B-cell ALL) are HLADR and CD 10 positive, which indicates a good prognosis. Infantile ALL : often high WBC, massive visceromegalies, severe thrombocytopenia, high rate of CNS involvement, poor response to treatment , high rate of relapse in comparison with childhood ALL, particularly extramedullary relapse and CD 10 negative leukemia

Children with T-cell ALL are characterized by: older age (peak at 8 years of age) with a ratio of boys to girls of 4: 1 high initial leukocyte count mediastinal enlargement high proliferation rate and/or frequent extramedullary manifestation (initially and at relapse) testicular involvment

ALL TREATMENT The treatment of ALL is risk-adapted depending on the different individual biological factors of ALL (clinical manifestation, laboratory analysis of morphology, cytochemistry, immunology, molecular cytogenetic, etc. ) The treatment of ALL is subdivided into remission induction, consolidation with CNS prophylaxis and maintenance phase

Induction of Remission Elimination of leukemic cells by a combination of vincristine, prednisone and additional cytotoxic agents such as daunorubicin, doxorubicin, and Lasparaginase. Duration of induction treatment: 4– 5 weeks Rate of first remission in ALL: more than 90%

For prophylaxis of CNS leukemic disease intrathecal application of MHA before, during, and after remission has to be performed. The addition of preventive irradiation is probably necessary in children at high risk of ALL as determined in most studies

Consolidation Treatment Without continuation of treatment leukemia will reappear within weeks or months. When remission with normal hematopoiesis is achieved further intensive chemotherapy is necessary to reach a complete eradication of leukemic cells. Combinations of different cytotoxic drugs reduce the number of remaining leukemic cells and the development of resistance against particular chemotherapies

Maintenance Treatment Risk-adapted maintenance treatment of different duration prevents recurrence of ALL Duration of treatment is 2. 5– 3 years with daily 6 mercaptopurine and once weekly methotrexate, with or without reinduction treatment are commonly used.

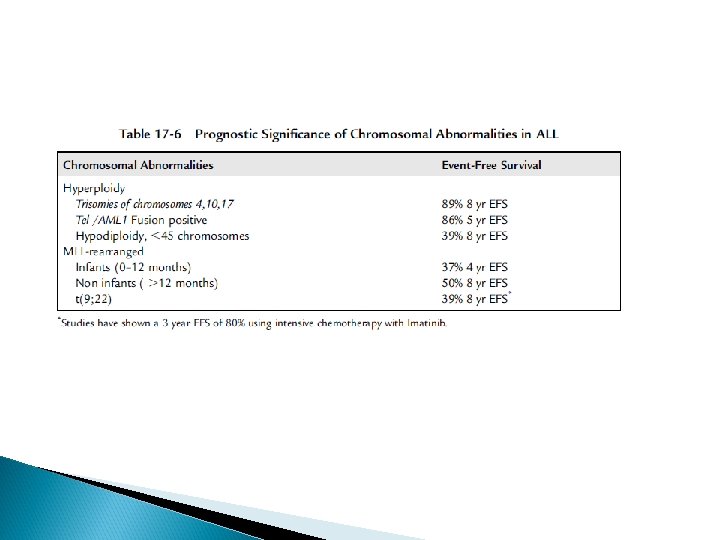
Philadelphia-Positive ALL Philadelphia-positive ALL is present in only 3– 5% of children with ALL. Previously less that 40% of these patients were cured with intensive chemotherapy. The use of imatinib TKI added to an intensive chemotherapy regimen has improved the outcome in this population at 3 years to an EFS of 80%.

Prognosis Approximately 80% of children with ALL survive without relapse with 7 - to 10 -year follow-up after diagnosis (long-term remission) In about one of five children a relapse of ALL occurs during maintenance treatment, within the first 6 months after treatment end or later. The risk of leukemia (recurrence) 5– 7 years after diagnosis is as low as in children without leukemia

Prognosis A relapse within the first 12 months after treatment stop indicates a poor prognosis A late relapse (more than 12 months after termination of maintenance treatment) usually has a better prognosis depending on the characteristics of the leukemic cells

Relapse Usually the same pheno- and genotype of ALL as at initial diagnosis. Rarely another cell line of leukemia ( a lineage switch) Differential diagnosis: secondary leukemia, which occurs years after initial diagnosis Intensive treatment necessary including hematological stem cell transplantation Event-free survival: after early relapse, 10– 30%; after late relapse, 40– 50%

HSCT INDICATIONS Induction failure: non remission BM at end of induction Philadelphia positive ALL Infantile ALL Hypodiploidy: less than 45 chromosomes Early isolated CNS relapse: less than 18 months from diagnosis Early BM relapse: relapse on therapy or less than 12 months after completion of therapy

QUESTIONS
- Slides: 46