Outline 1 what is BLAT why we need

Outline 1. what is BLAT & why we need it 2. BLAT's similarity & difference compared with BLAST 3. BLAT's application forms 4. BLAT's 3 major application 5. conclusion

What is BLAT & why we need it there exist many alignment tools -Smith. Waterman's algo : solves two short sequence alignment problem -FASTA, NCBIBLAST, Mega. BLAST WU-BLAST : provides flexible & fast alignment involving large database -Sim 4 : does a fine job with c. DNA alignment -SAM, PSI-BLAST : slowly but surely find remote homology

CONT process of assembling and annotating the human genome -aligning three millions ESTs and aligning 13 million mouse wholegenome random reads against the human genome -need to be done in less than two weeks in order to have time to process an updated genome every month or two ==>we need a very high speed alignment algorithm so the author developed BLAT the Blast-Like Alignment Tool
 -more accurate -500 times faster in m. RNA/DNA alignment")
CONT -BLAT(compared with existing tools) -more accurate -500 times faster in m. RNA/DNA alignment -50 times faster in protein/protein alignment -BLAT’s steps 1. using nonoverlapping k-mers to create index 2. using index to find homologous region 3. aligning these regions seperately 4. stiches these aligned region into larger alignment 5. revisit small internal exons possibly missed in first stage and adjusts large gap boundaries that have canonical splice sites where feasible
")
CONT -BLAT’s speed & sensitivity are decided by 1. k-mer size (finding hits step) 2. mismatch scheme (aligning step) 3. number of required index matches (find hits step)
 ie. build")
BLAT's similarity & difference compare with BLAST Similarity: -scans relative short matchs(hits) ie. build index then find hits -extend hits into high-scoring pairs (HSPs)

CONT Difference: -BLAST build index for query sequence but BLAT build index for database -BLAST scans linearly through database but BLAST scans linearly through query sequence -BLAST triggers an extension when one or two hits occur in proximity to each other but BLAT can trigger extensions on any number of perfect or near-perfect hits

CONT Difference: -BLAST returns each area of homology between two sequence but BLAT stitches them together into a larger alignment -BLAT has special code to handle introns in RNA/DNA alignments i. e. BLAT unsplices m. RNA onto the genome

BLAT's application forms server-client -building index is a relatively slow procedure a BLAT server is available for keeping index in memory for clients to query ==>good for interactive applications stand-alone -suitable for batch runs on one or more CPUs

BLAT's 3 major application & evaluation -m. RNA/DNA alignment -Mouse/Human Translated alignment -client/server version to power interactive searches
 -test set: remapped 713 m.")
CONT Evaluating m. RNA/DNA Alignments (compared with Sim 4) -test set: remapped 713 m. RNAs to genes on chromosome 22 -speed: BLAT: 26 sec Sim 4: 5 hr -sensitivity: BLAT: 99. 99% agreed of the annotated bases Sim 4: 99. 96%
 -for Human/Mouse : it has been")
CONT Evaluating Mouse/Human Tanslated Alignment (compared with TBLASTX) -for Human/Mouse : it has been shown that gapless alignment are in many ways preferable to gapped alignment for detecting coding regions
 speed comparison: method k N matrix")
CONT Evaluating Mouse/Human Tanslated Alignment (compared with TBLASTX) speed comparison: method k N matrix WU-TBLASTX 2736 s WU-TBLASTX 2714 s BLAT 5 1 +15/-12 5 1 BLOSUM 62 5 4 1 2 +2/-1 k: the size of perfect matching hit N: how many hits required to trigger a detailed alignment matrix: scoring method time 61 s 37 s
 sensitivity comparison: method Refseq exons WU-TBLASTX 84.")
Evaluating Mouse/Human Tanslated Alignment (compared with TBLASTX) sensitivity comparison: method Refseq exons WU-TBLASTX 84. 5% BLAT 86. 7% %chr 22 % Refseq Enrichment % bases 2. 67% 81. 7% 31 x 2. 89% 80. 8% 28 x %chr 22: percentage of chromosome 22 coverd by the alignment %Refseq: percentage of bases inside of human Ref. Seq coding sequence covered by the alignment Enrichment: column 2 / column 1 and high level indicate more specificity

CONT server/client to power interactive searches -thousands of interactive sequence searches per day -just one time for building index and keeps index in memory for query ===>efficient -but not as efficient as stand-alone version -because server need to save memory so it only keep the index, not the database

BLAT – The BLAST-Like Alignment Tool W. James Kent Genome Research 2002 陳韋仰

Database & Query Sequence Database : nonoverlapping Query sequence : overlapping database ……… K-mer query sequence K-mer

Three Search Criteria Single Perfect Matches Single Almost Perfect Matches Multiple Perfect Matches

Definition K : The K-mer size M : The match ratio between homologous areas H : The size of a homologous area G : The size of the database Q : The size of the query sequence A : The alphabet size 20 for amino acids 4 for nucleotides

T • How many nonoverlapping K-mers in the homologous region ? H : Homologous area size K : K-mer size

Single Perfect • : The probability that a specific K-mer in a homologous region of the database matches perfectly with the corresponding K-mer in the query = (M : The match ratio between homologous areas)

Sensitivity • P : The probability that at least one nonoverlapping K-mer in the homologous region matches perfectly with the corresponding K-mer in the query P = 1 – (1 – )T )T (T : #nonoverlapping K-mers in the homologous region)

Specificity • F : The number of nonoverlapping K-mers are expected to match by chance F = (Q - K +1) * ( #K-mers in the query sequence )*( #K-mers in the database )K that
 M P H = 100 ; G = 3 billion ,")
Single Perfect (Nucleotide) M P H = 100 ; G = 3 billion , Q = 500

Single Almost Perfect One letter may mismatch : The probability that a nonoverlapping K-mer in a homologous region of the database matches almost perfectly with the corresponding K-mer in the query = +K* (1 – M)

Sensitivity • P : The probability that any nonoverlapping K-mer in the homologous region matches almost perfectly with the corresponding K-mer in the query P = 1 – (1 – )T

Specificity • F : The number of nonoverlapping K-mers that are expected to match by chance F = (Q - K +1) * ( )*( )K + ) * (K * ( )K-1(1 - ( )))
 H = 100 ; G = 3 billion , Q")
Single Almost Perfect (Nucleotide) H = 100 ; G = 3 billion , Q = 500

Multiple Perfect • There must be N perfect matches, each no further than W letters from each other in the target coordinate, and have the same diagonal coordinate • Example : N = 2

Sensitivity • N=1, = • Pn : The probability that there are exactly n matches within the homologous region Pn = n(1 – )T – n ( ) • The probability that there are N or more matches => Pn+1 +…+PT

Specificity • FN : the number of chance matches of N K-mers each separated by no more than W from the previous match • N = 1, F 1 = (Q - K +1) * ( )*( )K
 • S : The probability of a second match occuring within W")
Specificity (continued) • S : The probability of a second match occuring within W letters after the first S = 1 – (1 - ( )K)W/K => Consider the Nth match is within W letters after the (N-1)th match FN = S * FN-1 FN = F 1 * SN-1
")
Multiple Perfect (Nucleotide)

Default Match Criteria Nucleotide : two perfect 11 -mer Protein : • stand-alone --- single perfect 5 -mer • client/server --- three perfect 4 -mer Reference : http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/BLAT_final. ppt

Implementation mickey

Algorithm 1. Search stage The program detects regions of the two sequences which are likely to be homologous. 2. Alignment stage Examining these regions in more detail and producing alignments for the regions.

Search stage 1. building up an index creating non-overlapping k-mers and their positions in the database. 2. excluding useless k-mers deleting K-mers that occur too often from index and containing ambiguity codes.

Search stage database … non-overlapping K-mer database position P 1 P 1 Index P 2 …
 P 1 P 1")
Search stage query sequence … overlapping K-mers database position (Kmers) P 1 P 1 … … Index P 2

Search stage Hit list K-mer P 1 database position P 1 query sequence position …
 picture from: http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/")
Search stage (example) picture from: http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/
 According to previous page, we know that… If diagonal values are")
Search stage (example) According to previous page, we know that… If diagonal values are equal, they are on the same diagonal. Position (query position, database position) Diagonal (DP-QP) aat 0, 3 +3 cac 6, 0 -6 cac 6, 9 +3 K-mer

Search stage DP – QP = 0 DP – QP < 0 query Coordinate DP – QP > 0 database Coordinate … bucket 1 bucket 2 bucket 3

Search stage Don’t care 2. Hits within proto-clumps are then sorted along the database coordinates and put into real clumps if they are within the window limit. 1. Hits that are within the gap limit are bundled together into proto-clumps. picture from: http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/

Search stage 1. 2. 3. Clumps with less than the minimum number of hits are discarded The rest are used to define regions of the database which are homologous to the query sequence. Clumps which are within 300 bases or 100 amino acids in the database are merged together. 500 additional bases are added on each side to form the final homologous region.

Search stage 3. Homologous region 1. Two clumps with the distance < 300 2. Adding 500 bases on each side picture from: http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/

Nucleotide Alignments 1. Search hits generating a hit list between the query and the homologous region of the database. 2. Extend hits
 The extension first merges adjacent hits and expands their ends")
Nucleotide Alignments (Extend hits) The extension first merges adjacent hits and expands their ends as far as the c. DNA and genomic DNA match perfectly. (overlapping hits are also matched) Allow N's in the c. DNA to match any single base. unaligned areas http: //www. soe. ucsc. edu/~kent/intronerator/algo. html
 The program then recurses, making up tiles and trying to")
Nucleotide Alignments (Extend hits) The program then recurses, making up tiles and trying to match in the unaligned areas. The recursion runs until either no tiles are found or until the gap between aligned blocks in the genome or c. DNA becomes less than 6 (5 in BLAT) Possibly introns tiles (Using smaller k to find http: //www. soe. ucsc. edu/~kent/intronerator/algo. html match in BLAT)

Nucleotide Alignments 1. Extensions that allow 1 or 2 mismatches if followed by multiple matches. 2. Extensions that allow 1 or 2 insertions or deletions (indels) followed by multiple matches are pursued. http: //www. soe. ucsc. edu/~kent/intronerator/algo. html

Protein Alignments The hits from the search stage are kept and extended into HSPs where a match +2 / mismatch 1 picture from: http: //www. csie. ntu. edu. tw/~kmchao/seq 05 fall/

Protein Alignments weight = B – Gap penalty = +25 HSPs B +45 query Coordinate Gap penalty = 20 (based on the distance between A and B) HSPs A +32 database Coordinate

Protein Alignments HSPs B +20 query Coordinate HSPs A +26 select “crossover” point to maximize the sum of the score of A up to the point and B starting at the point. database Coordinate

Protein Alignments A dynamic program then extracts the maximalscoring alignment by traversing this graph. The HSPs in the maximal-scoring alignment are removed, and if any HSPs are left the dynamic program is run again.

Stitching and Filling in Stitching alignments together using the same algorithm used to stitch together protein HSPs Multiple homologous regions

CONCLUSION -BLAT is a very effective tool for doing nucleotide alignments between m. RNA and DNA in same species -it is more accurate and faster than Sim 4 -BLAT's strategy for nucleotide alignments becomes less effective below 90% sequence identity but it can efficiently sequence divergence introduced by sequencing error

CONT High speed alignment program: two major stage 1. search stage: identify regions likely to be homologous -an index of some sort is key 2. alignment stage: does detailed alignments of the previously defined homologous regions.

CONT For search stage: -BLAT indexes database rather than query sequence so it only scan the short query sequence -A program “SSAHA” also indexes the database and it is an extremely effective tool for aligning genomic regions from same organism against each other -but “SSAHA” does not implement “unsplicing”, and always uses a single perfect match as a seed BLAT is more flexible in this aspect

CONT For search stage: -challenge to indexing is twofold: 1. size of index 2. time to generate index -size is not a problem because recently memory size is affordable -index is not generated often --->good in both batch or server/client mode

CONT For alignment stage: How index is used is important for speed -triggering an alignment for each matching is not always the optimal strategy to the index -mutiple near-by matches or longer k-mers but tolerating a mismatch both have much specificity for sensivity than one single match(BLAT implement a quick algo on this )
 Roger Yang 楊伍隆")
Human-Mouse Alignments with BLASTZ (Schwartz el al. 2000) Roger Yang 楊伍隆

Source S. Schwartz, W. J. Kent, A. Smit, Z. Zhang, R. Baertsch, R. C. Hardison, D. Haussler, and W. Miller Human-Mouse Alignments with BLASTZ Genome Research, 2003; 13: 103– 107

Mouse Genome Analysis Consortium Goals Study mutation and selection that shape the mouse and human genomes Estimating the fraction of the human genome under selection Determining the degree of regions under selection Measuring regional variation in the rate and pattern of neutral evolution

Sensitivity and Specificity Sensitivity is needed for aligning large portion of neutrally evolving genomic regions.

Homology Orthologs: two similar genes in two different species that originated from a common ancestor Paralogs: a gene in an organism is duplicated to occupy two different positions in the same genome

Software Design Issues Algorithm selections Have a high sensitivity requirement for detecting neutrally evolving DNA Fast algorithms (e. g. BLAT) sacrifice sensitivity for speed BLASTZ, used by Pip. Maker, is selected for the study.

Algorithm Evolution Gapped BLAST: specifically designed for aligning two long genomic sequence BLASTZ: another implementation of Gapped BLAST Modified BLASTZ: aligning entire mammalian genomes and better sensitivity

Software Design Issues BLASTZ Same 3 -step concept as Gapped BLAST ① Find short near exact matches ② Extend each short match without allowing gaps ③ Extend each gap-free match that exceeds a certain threshold by a dynamic programming procedure that allow gaps

Software Design Issues BLASTZ v. s. Gapped BLAST Option to require the matching regions must occur in the same order and orientation in both sequences A C G T Nucleotide 91 -114 -31 -123 1 A substitution matrix C -114 100 -125 -31 G -31 -125 -100 -114 -123 -31 -114 91 T 1. Chiaromonte et al. (2002)

Software Design Issues New BLASTZ speed improvement Regions are dynamically masked if several other regions are mapped to them. (e. g. zinc fingers or olfactory reception genes on Chromosome 19) Instead of 8 exact consecutive match nucleotides, use 1101001010111 as the space seed 1 To increase sensitivity, we allow a transition (A-G, G -A, C-T, T-C) in any one of the 12 positions 1. Ma et al. (2002) Pattern. Hunter

Software Design Issues BLASTZ algorithm 1. Remove lineage-specific interspersed repeat from both sequences. (Use Repeat. Masker for mouse and human) Lineage-specific: after the human-mouse split Interspersed Repeat: non-functional copy of RNA genes inserted into genome by reverse transcriptase. SINE (Short Intersperse sequences) – few hundred bp, about 11% of the human genome LINE (Long Intersperse sequences) – few thousand bp, about 21% of the human genome

Software Design Issues BLASTZ algorithm 2. For all pairs of spaced 12 -mers (one from each sequence) that are identical except perhaps for one transition, do the following. 2. 1 Extend the induced alignment in each direction without gaps. 2. 2 If the gap-free alignment scores more than 3000 then 2. 2. 1 Repeat the extension step with gaps. 2. 2. 2 Retain the alignment if it scores above 5000.

Software Design Issues BLASTZ algorithm 3. Between each pair of adjacent alignments from step 2, repeat step 2 use a more sensitive seeding procedure (e. g. 7 -mer exact match) lower score threshold: gap-free for 2000 (instead of 3000), gapped for 2000 (instead of 5000).

Software Design Issues BLASTZ algorithm 4. Adjust sequence positions in the resulting alignments to make them refer to the original sequences (i. e. , account for Step 1).

Software Design Issues BLASTZ algorithm 5. Filter the alignments Use axe. Best to filter out paralogs from orthologs Paralogs have greater sequence identity in a smaller region. Orthologs are usually longer with greater sequence identity.
 Mouse (2. 5")
Implementation and H/W Issues Segmentations Running time (with 888 Mhz PIII) Mouse (2. 5 Gb) into ~100 30 MB segments Human (2. 8 Gb) into ~3000 1. 01 MB segments (10 kb overlap) 481 days 12 hours with 1024 PC Output 9 Gbyte (relative position) Axe: 2. 5 Gbyte (actual bases) 3. 3% of the human genome is covered by multiple alignments

Software Evaluation Accuracy? Protein alignments are checked against X-ray crystal structures Gene predictions are checked against c. DNA library There’s no gold standard to verify genome alignment The easier to implement, the better

Software Evaluation Speed up options Early in the project, with only unassembled reads available, all-vs-all is the only option With all the genome sequence known, comparison can be made between smaller regions of human and mouse

Software Evaluation Specificity Conservation of synteny: Human chromosome 20 is considered to be completely homologous to parts of Mouse chromosome 2
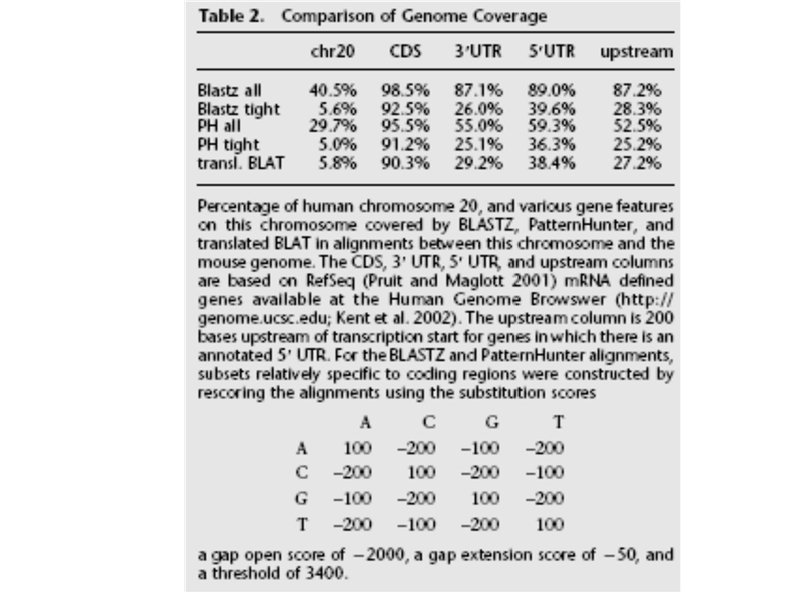

Discussion BLAST, BLAT, and Pattern. Hunter are tuned to align protein coding regions, not for fine-scale features of genome evolution 2 BLASTZ design philosophy Intended for all stage of sequencing Do not enforce critical a priori assumptions about which alignments are important; tasks of processing and filtering the initial alignments are left to other flexible programs
- Slides: 82