Otros nombres o Deficiencia fenilalanina hidroxilasa o Deficiencia


Otros nombres: o Deficiencia fenilalanina hidroxilasa o Deficiencia PAH

HISTORIA o El descubrimiento de la Fenilcetonuria por Folling en 1934 fue la primera demostracion de que este defecto genetico podia causar retraso mental.
 es una enfermedad del metabolismo,")
CONCEPTO o La fenilcetonuria o PKU (del inglés “phenylketonuria”) es una enfermedad del metabolismo, (autosomica recesiva) como consecuencia de una deficiencia de la fenilalanina hidroxilasa, una enzima que cataliza la hidroxilacion de fenilalanina a tirosina
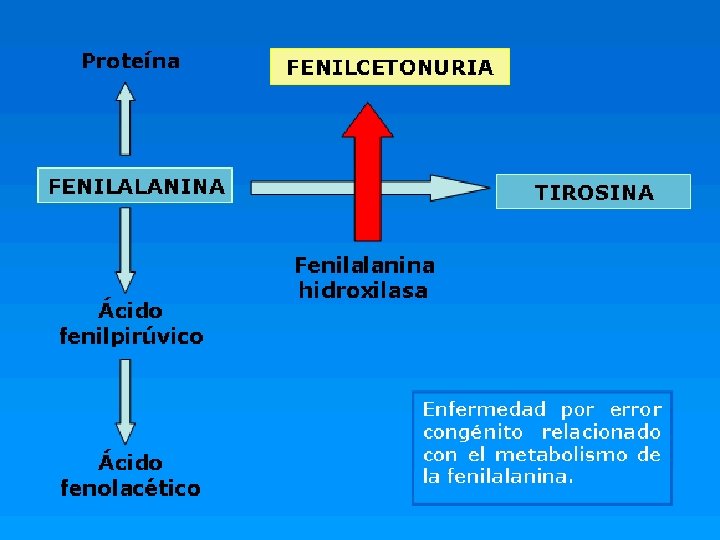

Consecuencia de NO Dx a tiempo. o La fenilalanina es uno de los ocho aminoácidos esenciales encontrados en los alimentos que contienen proteínas. o En este trastorno, la fenilalanina no se puede emplear en forma normal debido a la ausencia de la enzima, como consecuencia, se presenta una acumulación en el cuerpo de altos niveles de fenilalanina (hiperfenilalaninemia). o Estos compuestos son tóxicos para el sistema nervioso central y ocasionan daño cerebral.

ETIOLOGIA o La enfermedad se debe a una afectación del brazo largo del cromosoma 12: en el locus 12 q 24. 1

INCIDENCIA o En los EE. UU. , al menos uno de cada 25, 000 bebés nace con PKU. o En Europa es de 1/10, 000 nacidos vivos. o 1/20, 000 en Chile. o 1/6700 en Argentina (1998) n n 1/14500 PKU clásica 1/12000 PKU moderada

EPIDEMIOLOGIA o Este trastorno se produce en todos los grupos étnicos, si bien es más común en las personas cuyos antepasados provienen del norte de Europa o fueron indígenas nativos de los EE. UU. que en personas de origen afroamericano, hispano y asiático.
 o Microcefalia o Temblores o Movimientos espasmódicos de brazos")
SINTOMAS o Erupción cutánea (eccema) o Microcefalia o Temblores o Movimientos espasmódicos de brazos y piernas (espasticidad) o Postura inusual de las manos o Convulsiones o Hiperactividad o Retardo de las habilidades mentales y sociales o Retardo mental o Un olor distintivo de "ratón" en la orina y el sudor o Coloración pálida (es frecuente el cutis pálido, cabello rubio y ojos azules)

Alteraciones Fenilalanina Tirosina Ác. fenil láctico Acido fenilpirúvico Feniletilamina Ác. fenil acético Ác. p-hidroxifenil pirúvico

Manifestaciones clínicas Fenilalanina Alteraciones metabólicas en el cerebro Inhibición de la síntesis de serotonina Inhibición de la síntesis de melanina

Déficit de tirosina Tirosina Disminución de la síntesis de catecolaminas Disminución de la síntesis de tiroxina Disminución de la síntesis de tiramina

o Debido a que la fenilalanina está comprometida de forma indirecta en la producción de la melanina, el pigmento responsable del color de la piel y del cabello, los niños con fenilcetonuria usualmente tienen un cutis más claro que el de los hermanos no afectados.


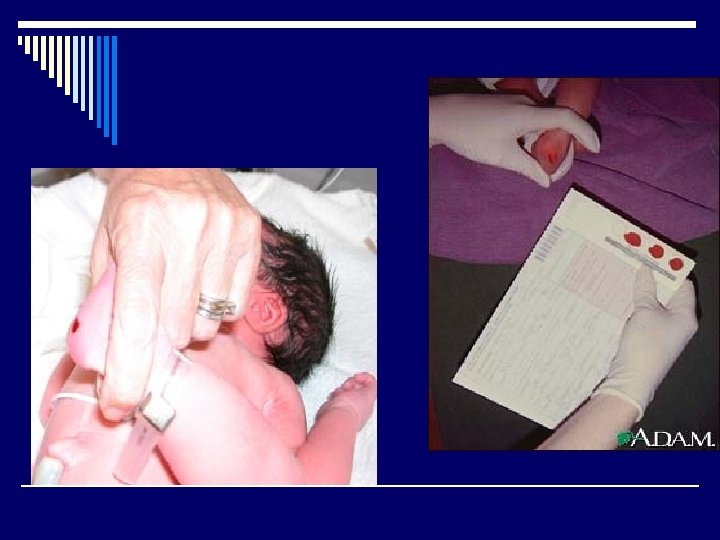
EXAMENES Y DIAGNOSTICO o Algunos de los exámenes que se realizan son: o Análisis enzimático para detectar el estado de portador (en los padres) o Prueba de las vellosidades coriónicas para detectar fenilcetonuria fetal (diagnóstico prenatal) o Tamizaje para fenilcetonuria (muestra de sangre del talón del bebé, extraída con una lanceta, para tamizaje y que es obligatorio en la mayoría de los estados de los Estados Unidos)

o En todos los estados y territorios de los EE. UU, se realizan pruebas de PKU a los bebés antes de que salgan del hospital. Ésta fue la primera prueba de detección precoz para neonatos del país. Se administra de manera rutinaria desde los años 60. Ya ha salvado a miles de niños del retraso mental.

o Se practica una pequeñísima punción en el talón del bebé para extraer unas pocas gotas de sangre. (La misma muestra puede utilizarse para detectar otros errores congénitos en la química del organismo. ) Por lo general, se envía la muestra a un laboratorio médico regional para determinar si la cantidad de fenilalanina es mayor a la normal y los resultados se envían al médico a cargo del cuidado del bebé. Si los resultados no son normales, se procede a la realización de pruebas adicionales para determinar si el bebé tiene PKU o si su alto nivel de fenilalanina se debe a alguna otra causa.


PKU MATERNA o 3, 000 mujeres en edad fecunda que han sido tratadas exitosamente de PKU en los EE. UU. La mayoría de ellas abandonó su dieta especial durante la niñez, porque en esa época la mayoría de los médicos creía que hacerlo no conllevaba riesgos. o Cuando estas jóvenes quedan embarazadas mientras o o comen una dieta normal, la concentración de fenilalanina en su sangre es muy elevada. Los niveles elevados de fenilalanina en la sangre materna durante el embarazo pueden causar problemas serios al feto. 90% de estos casos, los bebés sufren retraso mental. 70% por ciento nace (microcefalia). Muchos de ellos también nacen con defectos cardíacos y bajo peso.

TRATAMIENTO o Se alimenta al bebé utilizando una fórmula especial que contiene proteínas, pero sin fenilalanina. o Sólo se le administra leche materna o fórmula para bebés en pequeñas cantidades, para no darle más fenilalanina de la que necesita y es capaz de tolerar.

o Las personas con PKU deben seguir una dieta restringida durante toda su niñez y adolescencia y, en general, durante toda su vida (aunque es posible flexibilizar un poco la dieta en algunos casos con el avance de la edad)

ASPECTO PSICOSOCIAL Y PRONOSTICO DE VIDA o Las personas que presentan este defecto, es necesario como se menciono hacer la detección oportuna para iniciar el tratamiento lo mas rápido posible y evitar daño neurológico. o Deben estar informados que alimentos pueden consumir para evitar hiperfenilalaninemia.

FENILCETONURIA Alimentos ricos en fenilalanina • Leche materna • Leche de vaca • Productos lácteos • Carne • Pescado • Huevos • Aspartamo • Coca Cola (de todos los tipos) Nutra sweet o Equal (estos son edulcorantes artificiales que tienen fenilalanina) Alimentos pobres en fenilalanina • Vegetales • Frutas • Legumbres

FENILCETONURIA “Única enfermedad que presenta deficiencia mental, que puede ser
- Slides: 26