OTOZOMAL KROMOZOM HASTALIKLARI Kromozomal hastalklar Kromozomlarn bir btn

OTOZOMAL KROMOZOM HASTALIKLARI

Kromozomal hastalıklar: *Kromozomların bir bütün olarak veya bir parçalarının artması ya da azalması sonucu oluşurlar. • Otozomal ya da cinsiyet kromozomlarında bozukluk olabilir. * Kromozomlar onbinlerce gen içerdikleri için onlardaki ufak bir bozukluk bile fenotipik olarak belirgin değişikliklere yol açar. Taşıdıkları genlerin özelliklerine göre farklı fenotiplere yol açabilirler.

*Kromozomal hastalık, mental retardasyon, gelişme geriliği çeşit somatik anormallikle birliktedir. kromozomal hastalık sıklığı; • Yenidoğan dönemi %0. 7 • Ölü doğum %10 • çocukluk çağında ölüm %2. 5

• Kromozom hastalıkları etkisini büyük oranda doğumdan önce göstermektedir. • Trizomi ve monozomiler genellikle yaşamla bağdaşmaz • kendiliğinden(spontan) düşük ve gebelik kayıplarının önemli nedenidirler.

Spontan abortus olgularında kromozom anormallikleri Trizomi 13 Trizomi 16 Trizomi 18 Trizomi 21 Diğer kromozom trizomileri Monozomi-X Triploidi Tetraploidi Diğer %2 %15 %3 %5 %20 %15 %5 %10 Tanımlanabilen tüm Spontan abortusların %50 -60’ında neden Kromozomal anomalilerinden biridir.

Kromozom hastalıkları görülme olasılıkları Gebeliğin ilk üç ayında gerçekleşen spontan abortuslarda %50 -60 Geç gebelik kayıplarında ve ölü doğumda %5 Spermatositlerde %10 Oositlerde %25 Canlı doğumlarda %0. 7 35 yaş üzeri gebeliklerde %2

Kromozom bozukluklarının doğum öncesi ve sonrası yaşamın farklı dönemlerindeki görülme sıklığı Anormal karyotip ilk trimester düşükleri 35 Yaş üzeri annelerin fetusları Canlı doğumlar Toplam görülme sıklığı %50 -60 1/50 1/160 Sayısal anomali %96 %85 %60 Yapısal dengeli - %10 %30 %4 %5 %10 Yapısal dengesiz

Kromozom analizi gerektiren durumlar 1. 2. 3. 4. 5. 6. 7. 8. 9. Mental retardasyon Multiple konjenital anomali Ölü doğum ve neonatal ölüm Fertilite- cinsiyet gelişim problemleri Tekrarlayan gebelik kayıpları Ailede kromozom bozukluğu öyküsü 35 yaş üzeri kadınların gebeliklerinde Bazı kanserler Kromozom kırık sendromları

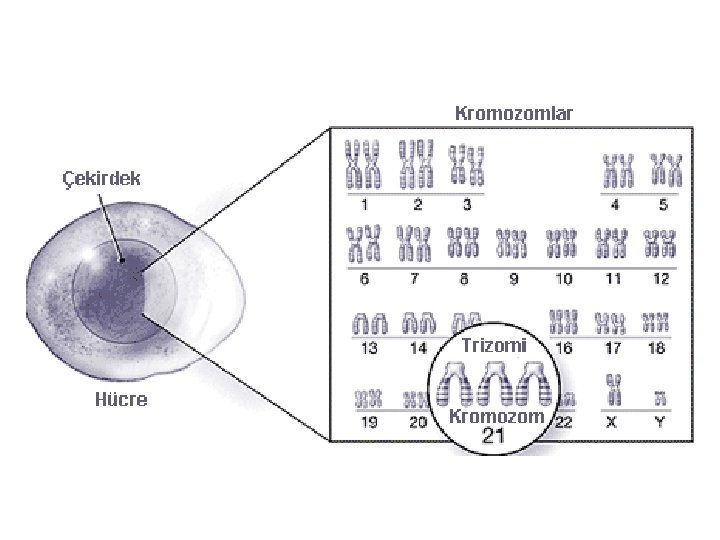
Sayısal kromozom bozuklukları • • 47, +13 47, +18 47, +21 45, X 47, XXY 47, XYY 47, XXX *triploidi *tetraploidi
")
Otozomal Trizomiler Postnatal yaşamla uyumlu olan yalnızca üç tane trizomi mevcuttur Trizomi 21(Down Sendromu) Trizomi 18 (Edwards Sendromu) Trizomi 13 (Patau Sendromu)

Down Sendromu 1866 J. Langdon Down klinik olarak tanımlamıştır 1959 Lejeune ve ark. Fazla bir 21. kromozom olduğunu saptamışlar En yaygın ve en iyi bilinen kromozom hastalığıdır. Orta derecede mental retardasyonun en sık genetik sebebidir. Sıklık 1/ 800 Tüm trizomi 21 gebeliklerinin sadece %20 -25’i doğuma kadar yaşayabilir.

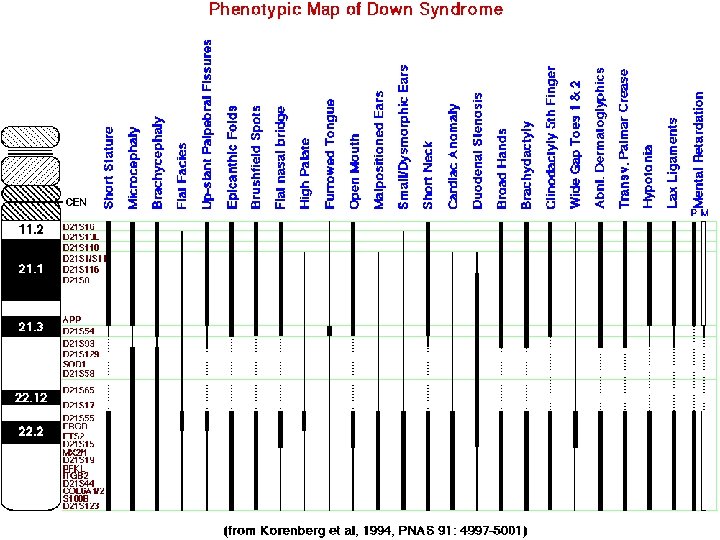
Fenotipik Bulgular • Brakiosefali • Hipotoni • Kısa boy • Kısa ve geniş eller • Gözlerde Brushfield noktaları • Sıklıkla tek bir elde palmar kriz (Smian Çizgisi) • Dismorfik Yüz görünümü: Burun kökü basık çekik gözler düşük yerleşimli ve kıvrık kulaklar ağız açık Protüzyone dil • Doğumsal kalp defekti gibi yapısal gelişim kusurları • Lösemi riskinde artış • Zeka oranı (IQ): %25 -50
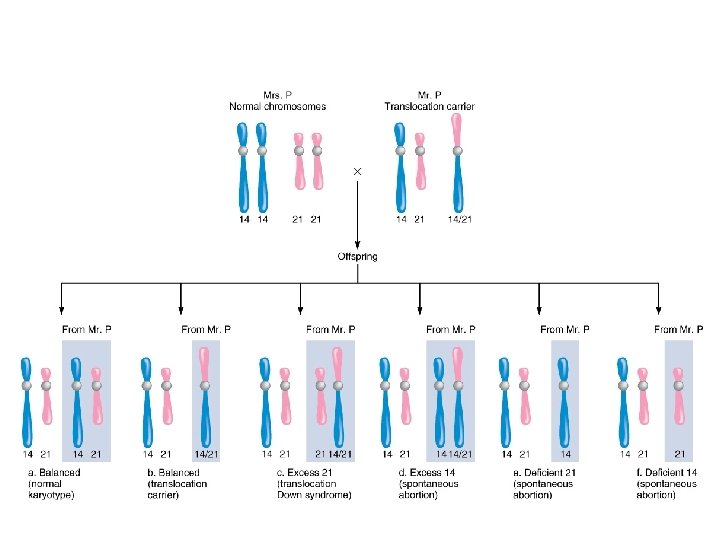
 %95 Robertson tipi translokasyon 46, rob(14; 21),")
Sitogenetik bulgular: Regüler Trizomi 21 (47, +21) %95 Robertson tipi translokasyon 46, rob(14; 21), + 21 %4 Mozaisizm 47, +21 / 46 %1 Regüler tipte fazladan 21 kaynağı %95 anne %5 baba
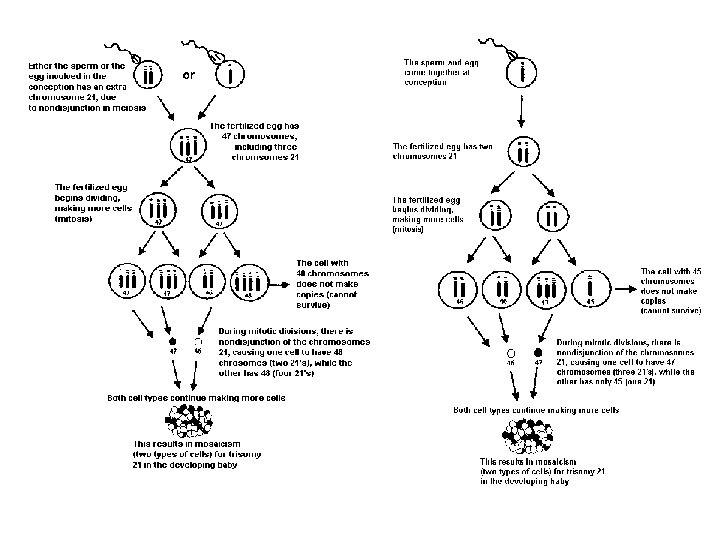

Down Sendromu Riski Hastalığın riski regüler tip DS için anne yaşına bağlı olarak artar; Sıklık 800 canlı doğumda 1’ ken 35 yaşında anne için risk 350 de 1’ e çıkar. 40 yaşında ise 100 de 1 dir. Translokasyon ve parsiyel trizomiler için anne yaşı risk oluşturmaz t(21; 21) taşıyıcısı için Down sendromlu çocuk doğumu olasılığı %100 dür!
 riski Ailenin Down Sendromlu bir çocuğu varsa ; sonraki gebelikte Down sendromlu")
Tekrarlama (rekürrens) riski Ailenin Down Sendromlu bir çocuğu varsa ; sonraki gebelikte Down sendromlu veya başka otozom trizomili bebek doğma riski %1 30 yaş altındaki annelerde bu risk %1. 4 30 yaş üstünde yaş riski ile aynıdır. üAilede başka bireyde DS bebek öyküsü riski arttırmaz.
 Baba")
Translokasyon tipi ile DS riskleri Translokasyon tipi Taşıyıcı Amniyosentezle saptanabilen DS rob(14; 21) Baba %1 rob(14; 21) Anne %15 rob(21; 22) Baba %5 rob(21; 22) Anne %10 rob(21; 21) Her iki eş %100
 Sıklık 7500 canlı doğumda 1’ dir. Tüm trizomi 18 lerin")
Trizomi 18 (Edwards Sendromu) Sıklık 7500 canlı doğumda 1’ dir. Tüm trizomi 18 lerin % 95 i spontan abortusla kaybedilir. Hastaların %80’ i kızdır. Mayoz 1 hatasından kaynaklanır. Hata %95 annede %5 babada oluşur Maternal yaş risk oluşturur.

Fenotip: Büyüme gelişme geriliği mikrosefali Belirgin oksiput Hipertoni Ellerde tipik fleksiyon Kalp anomalileri

Sitogenetik 47, XX, +18 Olguların % 8 i mozaiktir % 4 çifte anöploidi vardır 48, XXY, +18 48, XXX, +18 48, XX, +18, +21 veya +13 %20 -25 dengeli translokasyon taşıyıcısı bireyin dengesiz gamet dağılımı sonucu oluşur. Ailede bir çocuk hasta olduğunda tekrarlama riski %1. 5 dir
 Sıklık 20 000 -25 000 canlı doğumda 1’dir. Fenotip •")
Trizomi 13 (Patau Sendromu) Sıklık 20 000 -25 000 canlı doğumda 1’dir. Fenotip • Ciddi kalp ve santral sinir sistemi anormallikleri • Ellerde ayakta post aksiyel polidaktili • Yarık damak yarık dudak • Sıkılmış yumruk En fazla 6 ay yaşayabilirler. Çoğu antenatal dönemde kaybedilir.
, +13 46, rob(13; 15), +13 46, rob(13; 21),")
Sitogenetik 47, +13 46, rob(13; 14), +13 46, rob(13; 15), +13 46, rob(13; 21), +13 46, rob(13; 22), +13 46, t(13; ? ) Tekrarlama riski % 2 İleri anne yaşı risk oluşturur.
 Doğumdaki insidansı 1 yılın üzerinde yaşayan gebelikler % 13 (Patau) 1/12, 500")
Trizomi (sendrom) Doğumdaki insidansı 1 yılın üzerinde yaşayan gebelikler % 13 (Patau) 1/12, 500 - 1/21, 700 <5% 18 (Edward) 1/6, 000 - 1/10, 000 < 5% 21 (Down) 1/800 - 1/826 85%

Otozomal delesyon sendromları Dismorfik hastalarda sitogenetik olarak saptanabilen bir kaç delesyon bildirilmiştir. Bunlardan çok azı tanımlanmış belli bir sendromla birliktedir. 7000 canlı doğumda 1 sıklıkla sitogenetik olarak saptanabilen otozomal delesyonlara rastlanmaktadır. Mental retardasyon Dismorfik yüz

Wolf Hirschhorn Sendromu 4 p-
 ~15%")
G-banding 60% HRB 15% FISH 25% ~85% de novo delesyon” (85% paternal kromozomda) ~15% “ring 4” veya “ 4 p-mozaikliği”, “der 4” translokasyona bağlı dengesiz gamet (2/3 anne taşıyıcı) WH sendromlu çocukların anne babaları dengeli yapısal anormallik açısından incelenmeli.

Yeme problemi Nöbet geçirme Büyüme geriliği

Cri Du Cat Sendromu 5. Kromozomun kısa kolunda büyük bir delesyon vardır Böyle çocuklar ağlarken kedi miyavlaması şeklinde ses çıkarırlar Tüm mental retarde çocukların %1 de saptanmaktadır Mikrosefali (küçük baş) Hipertelorizm (birbirinde uzak göz mesafesi) Epikantal fold Düşük kulak Peri aurikular tag (kulak önü deri çıkıntısı) Mikrognati (küçük çene)
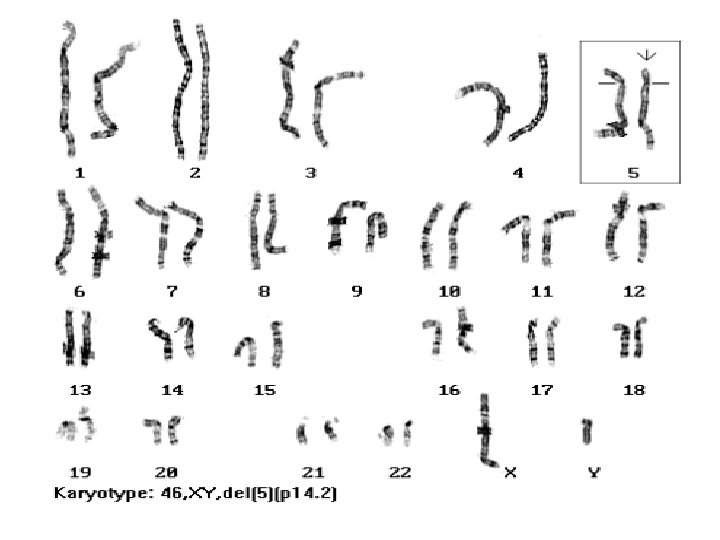

Vakaları çoğunluğu sporadiktir. %10 -15’ i 5. kromozomu içeren translokasyon taşıyıcısı anne/baba çocuğudur Tüm hastalarda 5. kromozomun kısa kolu aynı büyüklükte delesyona uğramaz. Fenotipik hastalarda 5 p 15 bandının kaybı ortaktır. Klinik bulguların çoğu 5 p 15. 2 bandındaki genlerin monozomisi sonucu oluşan gen haploinsuffisiensi nedeniyledir. 5 p 12. 3 bandı farklı özellikte Cat cry bulgularına neden olur.

Mikrodelesyon sendromları Bir kaç dismorfik sendrom küçük fakat bazen sitogenetik olarak saptanabilen küçük kromozom segmentlerinde delesyonla birliktedir. Bu delesyonlar klinik olarak saptanabilen farklı sendromlara neden olur. *Mikrodelesyon sendromları *Bitişik gen sendromları *Subtelomerik delesyon sendromları Yüksek rezolüsyon bantlama (HRB) veya Moleküler Sitogenetik yöntemlerle (FISH: Floresan insitu hibridizasyon) saptana bilirler.
 gen")
Delesyon bölgesinde bağlı genlerin bulunması, birlikte bulunan bu genlerin kaybı sonucu contiguous (bitişik) gen sendromlarına yol açar. Diğer delesyon sendromları oldukça geniş olan tek bir gendeki delesyonlardan kaynaklanmaktadır.

Subtelomerik bölge delesyonları mental retardasyon ve konjenital anomalilerle birlikte görülebilir. Tanımlanmış birkaç subtelomerik delesyon sendromu bulunmaktadır.

Her sendrom için farklı hastalarda aynı gene ait farklı büyüklükte delesyon ve bazende duplikasyon vardır. Mayoz 1 deki eşit olmayan krossing-over nedeniyle olduğu ileri sürülmektedir. Smith-Magenis sendromu 17 p 11. 2 dup(17 p 11. 2) delesyon duplikasyon Prader Willi/Angelman S 15 q 11 -q 13 delesyon Williams Sendromu 7 q 11. 23 delesyon İktiyozis Xp 22. 3 delesyon Nörofibromatozis 17 q 11. 2 delesyon Di. George /Velokardiofasial S 22 q 11. 2 delesyon Cat-eye sendromu 22 q 11 duplikasyon

Kromozom 22 q 11. 2 bölgesindeki gene ait Mikrodelesyon sendromları Velokardiofasiyal Sendrom Di. George sendromu Bu bölgedeki 3000 kb lik gen delesyonları neden olur 1/ 2000 -4000 sıklıkla görülürler VCFS MR Kraniofasial anomaliler Kalp defektleri Tüm konjenital kalp defektlerinin %5’inden sorumlular Bu gen bölgesindeki duplikasyon ise Cat-eye sendromuna yol açmaktadır.

22 qter 22 q 11. 2

Williams Sendromu 7 q 11. 23 delesyon Mavi gözler, yıldız şeklinde iris Belirgin dudaklar Acık ağız Uzun filtrum Kısık ses Kardiyovasküler sistem anomalileri IQ 40 -80
 Angelman Sendromu (AS) Babadan")
15 q 11 -q 13 delesyon sendromları Prader Willi Sendromu(PW) Angelman Sendromu (AS) Babadan geçen 15. kromozomda delesyon PW Anneden geçen 15. kromozomda delesyon AS Delesyon saptanmayan durumlarda uniparental dizomi (UPD) veya genomik imprinting UPD; Her iki 15 babadan gelirse AS Her iki 15 anneden gelirse PW

Prader Willi Sendromu Hipotoni Belirgin obesite Orta dereceli MR Kısa boy Küçük el-ayak Hipoplastik dış genital organlar Sıklık 1/ 1000 Neden: %50 babadan geçen 15 de saptanabilir delesyon %20 babadan geçen 15 de mikrodelesyon %25 UPD (her iki 15 anneden) %2 translokasyon taşıyıcı babanın dengesi gametogenezisi
 MR Koordinasyon bozukluğu")
Angelman Sendromu Gelişme geriliği Uygunsuz gülme atakları Kukla yüz (hapy puppet) MR Koordinasyon bozukluğu Sıklık: 1/20 000 Nedenleri: %50 saptanabilir delesyon (anneden geçen 15 de) %25 Mikrodelesyon %5 UPD (her iki 15 babadan geçer) %20 bilinmiyor
 Bloom Sendromu 15 q 26")
Sitogenetik olarak etkilenen Mendelian Hastalıklar (Kromozom kırık sendromları ) Bloom Sendromu 15 q 26 OR artmış SCE oranları Ataksi telenjiektazi 11 q 13 OR DNA tamiri bozuk, kromatid hasarı Fankoni Anemisi 16 q 24 OR artmış SCE oranları Kseroderma pigmentoza OR artmış SCE oranları Frajil X X’ geçişli Xq 27. 3 kırıkları Xq 27. 3 Roberts sendromu OR Sentromerik heterokromatin hatalı bölünmeleri ICF OR spesifik DNA bölgelerinin hipometilasyonu; 1, 9, 16 cen

DNA kırıkları ve kromozom SCE

FRAJİL X CGG tekrar artışı

Fankoni anemisi DEB testi

Roberts S

Kanser ve kromozom
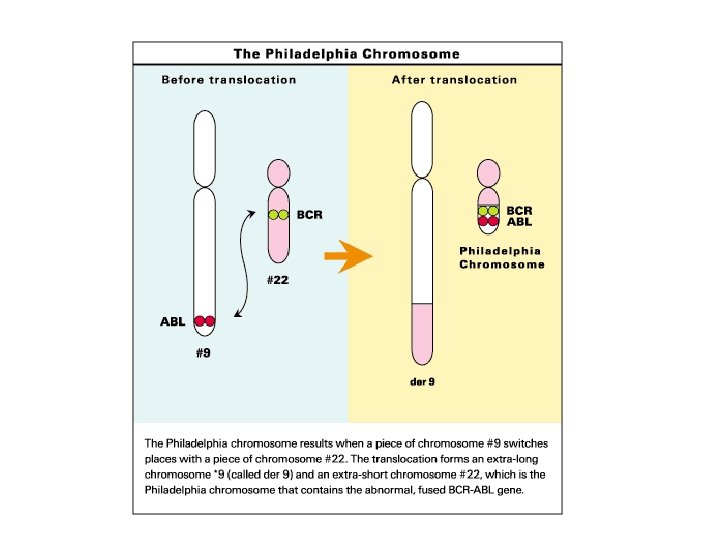


Double minute
- Slides: 53