or om c s t ren Rele ase



, fiziologice, biochimice şi comportamentale, observabile sau")







 → anomalii structurale v")







 n n Pot")





 la copii sunt diagnosticate în baza unor semne şi/sau trăsături")



, +18 Dolicocefalie Gât sucit Arcuri pe 3")


; trisomie")













 n n n incidenţă 1 la 1000 nn de sex")
 n datorită disgeneziei gonadice: n n n n n absenţa")





 n n Pot fi:")


 Terminală Interstişială")
 Paracentrică Pericentrică")
 Reciprocă Cu inserţie Robertsoniană (rob)")
 Cromozom inelar (r) Izocromozom (i, iso)")
")

 (q 11 -q 13), crs")


 – inactivarea selectivă a alelei în dependenţă de originea ei parentală")




 Copiii cu anomalii congenitale multiple (minore/majore) asociate cu:")

- Slides: 77
or om c. s t ren Rele ase ed. T M by
BOLI GENETICE reprezintă stări patologice determinate preponderent de factori genetici, ce apar ca o consecinţă a erorilor (mutaţiilor) la nivelul materialului ereditar. 1. 2. 3. Actualmente în Republica Moldova există un număr relativ mare de copii cu patologii genetice, generate de : anomalii cromosomiale numerice şi structurale boli monogenice boli multifactoriale - malformaţii congenitale izolate şi multiple etc.
Impactul major al frecvenţei acestor stări patologice în pediatrie se explică prin faptul că transmiterea ereditară, influienţa anumitor factori teratogeni pe parcursul perioadelor precoce de dezvoltare ontogenetică determină apariţia modificărilor genotipice şi fenotipice fatale la făt, iar ulterior la copilul nou-născut.
n Fenotipul reuneşte totalitatea caracteristicilor fizice (somatice, morfologice), fiziologice, biochimice şi comportamentale, observabile sau detectabile ale unui individ. El este rezultatul interacţiunii factirilor ereditari şi a factorilor de mediu. Genotipul reprezintă informaţia ereditară, materializată în genele conţinute în cromozomi şi care este constant în tot cursul dezvoltării ontogenetice. Fenotipul este potenţial variabil.
Genetica este ştiinţa eredităţii şi variabilităţii Ereditatea defineşte proprietatea unui individ de a transmite la urmaşi caracterele sale personale, precum şi cele de specie. Variabilitatea cuprinde fenomenele care produc diferenţele genetice dintre indiviziii unei populaţii şi dintre populaţii diferite.
Substratul material al eredităţii şi variabilităţii n Molecular n Morfologic
Realizarea informaţiei genetice n La nivel molecular - sinteza unei proteine Formarea unei proteine funcţionale ce asigură: - realizarea unei structuri celulare - unui lanţ metabolic - unei căi de semnalizare - realizarea unui caracter morfologic, fiziologic, biochimic. . . (structură anatomică, proprietate, funcţie specifică a unui organ. . . )
Genomul uman v v v termenul GENOM a fost introdus de Winkler în 1920, definind totalitatea materialului genetic al unei celule sau a unui individ. este format din unirea a 2 termeni: GENa şi cromoz. OM reprezintă lotul haploid de cromozomi sau totalitatea nucleotidelor situate în ADN-ul cromozomilor Genomul uman este format din 3 mlrd. perechi de baze azotate, sau 3 mln. De kilobaze, sau 3 mii de Megabaze. v cartografiere – det-rea poziţiei locilor în crm; v Secvenţare determinarea secvenţelor de baze nucleotidice
ă c i s a l c a i Relaţ Medic Abordarea genetică Pacient Familie
Particularităţile bolilor genetice: sunt determinate periconcepţional şi prenatal • pot fi: * congenitale, dar se pot manifesta la intervale ontogenetice diferite: - prenatal (anomalii de dezvoltare din partea SNC: hidrocefalie, spina bifida, defecte de tub neural; M. C. de cord; avorturi spontane; sarcini stopate în evoluţie etc. ) - neonatal (anomaliile cromozomiale: boala Down etc. ) - postnatal (unele anomalii cromozomiale (sindr. Turner, Klinefelter etc. ) şi boli monogenice: fenilcetonuria, distrofia musculară Duchenne, choreea Huntington, neurofibromatoza etc. , amiotrofiile spinale, M. C. de cord, renale, digestive - stenoza congenitală hipertrofică de pilor, hernia diafragmală etc. ) •
pot fi: - ereditare, se transmit din generaţie la alta, dar nu neapărat, deoarece pot să apară ca consecinţă a unei mutaţii spontane, noi - mutaţie “de novo”; - boli ce afectează reproducerea, ne fiind transmise la urmaşi (criptorhidia, hipoplazia uterului etc. ); - familiale, dar pot fi şi cazuri izolate, - nu toate bolile familiale sunt şi genetice (TBC, diverse infecţii). n evoluţia - cronică, progredientă, recidivantă. rezistenţa la metodele tradiţionale de n tratament.
Clasificara bolilor genetice v Boli cromozomiale: → anomalii numerice (aneuploidii) → anomalii structurale v v Boli monogenice: AD, AR, X - linkate, Y – linkate Boli mitocondriale: cu transmitere pe linie maternă sau în conformitate cu modurile mendeliene. Boli multifactoriale – cu predispoziţie genetică. Boli ale genomului celulelor somatice: se produc după concepţie, sunt limitate la celulele somatice şi NU se transmit la descendenţi.
SINDROAMELE CROMOZOMIALE !!! SUNT FRECVENTE !!! AU MANIFESTĂRI VARIATE !!! AU CONSECINŢE MAJORE
SINDROAMELE CROMOZOMIALE n n n - constituie un grup de patologii genetice la baza cărora stau mutaţiile cromozomiale; - reprezintă modificări genetice produse prin mecanisme cromozomiale specifice: segregarea anormală a cromozomilor în meioză sau în mitoză; recombinare intercromozomială aberantă; reparare greşită a rupturilor cromozomiale.
1 2 13 4 3 14 15 5 16 6 17 8 7 18 19 9 20 10 21 11 22 12 X Cariotip 46, XX diploid = 50% matern +50% patern
Clasificarea anomaliilor cromozomiale
Criteriu Tip de anomalie Momentul producerii - constituţională - dobândită Tipul de afectare a * numerică: materialului genetic - echilibrate - neechilibrate * structurală: - echilibrate - neechilibrate * disomie uniparentală (UPD) Număr de celule afectate - omogenă - în mozaic; himeră Tipul de cromozom afectat - autozomală - gonozomală Tipul de celule afectate - somatică - germinală
Sindroame cromozomiale de număr şi de structură n n Constituţionale (~0, 83% nn; >8% sarcini) n Neechilibrate (0, 4%) – fenotip anormal: n Echilibrate (0, 43% nn) – fenotip normal + tulburări de reproducere; Dobândite → cancere !!! Anomalii neechilibrate – poliploidii, aneuploidii, aberaţii cromozomiale neechilibrate (del, dup, i, r)
Anomalii cromozomiale constituţionale NEECHILIBRATE (0, 4% nn – 1: 250 nn) n n Pot fi : n an. de număr / structură n trisomii complete / parţiale (dup) n monosomii complete / parţiale (del); n omogene / mozaic Reprezintă anomalii de dozaj genic Se manifestă cu fenotip anormal: n Întârziere de creştere pre- şi postnatală; n ACM minore ± majore n RM ± tulburări comportament n Tulburări sexualizare / reproducere Prezintă gravitate diferită care depinde de: n mărimea dezechilibrului, n tipul anomaliei, n cromozomului implicat, n Nr. celule afectate
MECANISMELE DE PRODUCERE ALE POLIPLOIDIILOR n n ERORI DE MEIOZĂ – nesepararea citelor de ordinul II ERORI DE FECUNDARE n Diginia n Dispermia n Diandria n Endoreduplicarea
Nesepararea gametocitelor II
Erori la fecundare DISPERMIA DIANDRIA DIGINIA NE SEPARAREA GLOBULUILUI POLAR
MECANISMELE DE PRODUCERE ALE ANEUPLOIDIILOR ACCIDENT în cursul: FORMĂRII CELULELOR SEXUALE LA UNUL DIN PĂRINŢI n Nedisjuncţie cromozomială AI n Întârziere anafazică AI n Nedisjuncţie cromatidiană AII n Întârziere anafazică AII • PRIMELOR DIVIZIUNI ALE ZIGOTULUI • Nedisjuncţie cromatidiană • Întârziere anafazică n
NONDISJUNCŢIA MEIOTICĂ n n n Maternă 92% din trisomiile 21 Paternă 100% 47, XYY; 70% 45, X sau 47, XXY Cauze ? ? ? Factorii externi nu au nici un rol !!! n Efectul vârstei materne (sinapsa prelungită a cromozomilor omologi) – parţial !!! (75% din copii cu s. Down se nasc din mame tinere) n
Majoritatea sindroamelor genetice (cromozomilae) la copii sunt diagnosticate în baza unor semne şi/sau trăsături comune (aspectul fenotipic) ale manifestărilor clinice. În general, multe sindroame sau boli genetice, sunt rar întîlnite şi deaceea nu toţi medicii posedă aptitudini în diagnosticarea acestor afecţiuni. Chiar şi în secolul nostru există încă multe deosebiri de opinie a ce reprezintă un sindrom genetic. Cel mai des acestea se împart în două categorii (sau grupe): n Sindroame prin definiţie n Sindroame prin etiologie
n Sindromul prin definiţie constituie o asociere de defecte morfologice, fiziologice, structurale şi funcţionale observate de autorul care a descis primul patologia, iar sindromul respectiv poartă numele acestui autor (sindromul Rubinstein-Taybi „police lat şi haluce mare”, sindromul Cornelia de Lange etc. ). n Sindromul prin etiologie include toate combinaţiile de anomalii cunoscute de aceaşi etiologie (mutaţii cromozomiale, genice şi genomice: sindr. Down, Patau, Edwards; sindroamele monogenice – sindr. Lourence-Moon. Biedl, sindr. Marfan, sindr. Elers-Danlos etc. ).
Iniţial, multe sindroame cromozomiale erau descrise ca sindroame prin definiţie. Ex. : sindromul Turner era original recunoscut ca asocierea a 4 trăsături caracteristice: hipostatură, edem, gît palmat şi amenoree primară la sexul femenin. Ulterior, constatînduse cauza etiologică a acestei boli genetice cromatina sexuală negativă la fetiţe, determinarea unui cariotip 45, XO, sindromul Turner este considerat un sindrom cu o etiologie cunoscută.
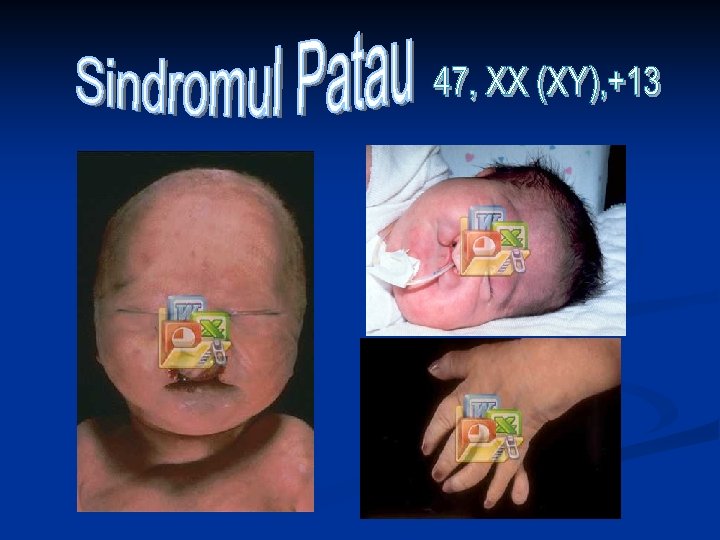
Retard fizic şi mintal Microoftalmie Urechi deformate, surditate Pliu palmar transvers unic Triradius axial distal Defectul septului inimii Inima – în partea dreaptă Rinichi polichistic Dublarea ureterelor Arc S-form pe talpă în regiunea halucelui Segmentare exagerată a nucleilor PMN 47, XX (XY), +13 ♀>♂ Microcefalie, arinencefalie Defecte ale craniului Hipotelorism Despicătura buzei şi palatului Polidactilie Anomalii ale unghiilor Hidronefroza Hernie ombilicală Uter divizat Criptorhism
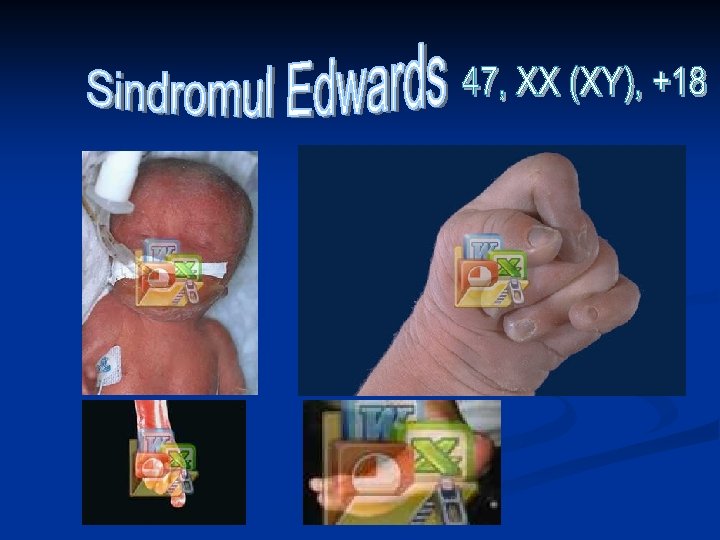
Retard fizic şi mintal 47, XX (XY), +18 Dolicocefalie Gât sucit Arcuri pe 3 sau mai multe degete Pliu palmar transvers unic Stern scurt Lipsa unei artere ombilicale Rinichi în formă de potcoavă Hipertonie musculară Flexie dorsală a degetului mare ♀>> ♂ Fontanele mari Hipertelorism Urechi deformate Micro- şi retrogtatie Anomalii de flexie a degetelor Defectul septului interventricular Diverticuli intestinali Anomalii multiple ale organelor genitale
SINDROMUL DOWN SAU TRISOMIA 21
SINDROMUL DOWN SAU TRISOMIA 21 n n n John Langdon Down – 1866; Jerome Lejeune – 1959 trisomia 21. Incidenţa 1: 700 nn; Simptomatologia aspect fenotipic caracteristic> n hipotonie musculară, hiperlaxitate articulară şi hiporeflexie nervoasă. n dismorfism cranio-facial sugestiv. n mâinile sunt scurte şi late, clinodactilie deget V şi, frecvent, pliu simian; n inconstant, malformaţii viscerale (defecte cardiace, atrezie duodenală, imperforaţie anală). n Întârziere în dezvoltare psiho-motorie / RM (QI variază între 20 şi 85).
ANALIZA CITOGENETICĂ în sdr. Down n n trisomie liberă şi omogenă (92 -95%); trisomie 21 liberă în mozaic cromozomic (47/46) (23% din cazuri); trisomie 21 prin translocaţie Robertsoniană neechilibrată n între cromozomul 21 şi un alt cromozom acrocentric – (4 -5% din cazuri); n frecvent t(21 q; 14 q), mai rar t(21 q; 22 q) sau t(21 q; 21 q); t(21 q; Dq) sunt moştenite în 50% cazuri, cel mai adesea de la mamă; t(21 q; Gq) sunt de obicei de novo. !!! în aceste cazuri se va face cariotipul ambilor părinţi
Identificarea numărului de cromosomi 21 în nucleii interfazici prin metoda FISH 46, XX 47, XX, +21 Cariotipul obţinut prin bandare G a unei femei cu sindrom Down
Carotipuri posibile în s. Down n n n 47, XX, +21 47, XY, +21 46, XX / 47, XX, +21 46, XY / 47, XY, +21 46, XX, rob(21; 21) 46, XY, rob(21; 14) 46, XY, i (21 q)
SINDROAMELE CU ANOMALII ALE CROMOZOMILOR SEXUALI n Anomaliile cromozomilor sexuali – au o incidenţă globală de: n n n 1 la 400 nn sex masculin (47, XXY; 47, XYY) 1 la 650 nn sex femenin (45, X; 47, XXX) Fenotipuri mai puţin severe; se asociază frecvent cu: n n n întârziere în dezvoltarea pubertară, disgenezii gonadice (amenoreea primară-secundară; azoospermia) sterilitate.
SINDROMUL TURNER n Disgenezie gonadică monosomia X, comletă sau parţială; omogenă sau în mozaic: 45, X; 46, XX/45, X; 46, X, i(Xp); 46, X, i(Xq); 46, X, Xp-; 46, X, Xq-; 46, X, r. X. . . n n 2% din concepţii, dar este letală în 95% din cazuri, 1/2500 -1/3000 din nou-născuţi de sex feminin.
Principalele aspecte clinice, sunt sugestive pentru Sindromul Turner, in dependenţă de varsta persoanei cu acest sindrom. * Perioada neonatală * Perioada prepubertară * Perioada postpubertară
Întarziere majoră în creştere, ce devine evidenta dupa 2 -3 ani, şi atinge la 10 ani – 3 DS (deviaţie standard) faţa de medie. Gat scurt, palmat cu inserţia joasă a parului pe ceafă. Torace lat cu mameloanele mult distanţate.
* Hipostatură * Amenoree primară * Caractere sexuale secundare feminine deficitare.
Persoanele îndreptate pentru diagnostic prenatal: Vârsta maternă > 35 ani Triplul test anormal: AFP – redus, estriol neconjugat – redus, HCG – crescut. Ecografie fetală suspectă pentru Sindrom Turner: hygroma cysticum, hidrops fetal, malformaţii congenitale cardiace, malformaţii congenitale renale
Reprezentarea schematică a taliei diferitor categorii de persoane.
Analiza citogenetică este decisivă pentru diagnosticul s. Turner n n n Testul cromatinei X este negativ; testul cromatinei X= test sceening simplu şi ieftin Examenul cromozomial – esenţial pentru diagnosticul de certitudine. monosomie X omogenă (50 -60%) ND paternă (70%); mozaicuri 45, X/46, XX ş. a. (25%); anomalii structurale cromozomului X: isocromozomi, deleţii, crs. inelari.
Evoluţie şi prognostic în s. Turner n n n Depistarea precoce – permite folosirea unei terapii eficace: n hormon de creştere (se câştigă 6 -10 cm la talia finală), n prepubertar, cu estrogeni, corijează deficitul de sexualizare fenotipică. probleme deosebite pun copiii cu sindrom Turner care au malformaţii cardiace sau renale – complicaţii. Ulterior: tiroidite autoimune, HTA, obezitate şi DZID. sterilitate Inserţia socială este de obicei bună, viaţa de familie este posibilă.
SINDROMUL KLINEFELTER (47, XXY) n n n incidenţă 1 la 1000 nn de sex masculin (probabil 1/600) Diagnosticul clinic este posibil doar postpubertar. multe cazuri nu sunt diagnosticate – modificări fenotipice reduse. talia creşte, pe seama membrelor inferioare. Penisul se dezvoltă, de obicei, normal şi funcţia sexuală este normală. Testiculii rămîn mici (sub 3 cm x 1. 5 cm), fermi, nedureroşi la palpare („disociaţie peno-orhitică”).
SINDROMUL KLINEFELTER (47, XXY) n datorită disgeneziei gonadice: n n n n n absenţa spermatogenezei – sterilitate primară şi definitiv; Absenţa testoteron caracterele sexuale secundare slab dezvoltate + valori crescute FSH şi LH. În circa 30% din cazuri apare ginecomastia Dezvoltarea intelectuală este aproape normală Speranţa de viaţă este normală. Pot apărea probleme de adaptare socială şi tulburări de comportament. Analiza citogenetică: cromatina X= pozitivă 47, XXY (85%), mozaicuri 46, XY/47, XXY sau polisomii XY (48, XXXY, 49, XXXXY) – 13%; un fenotip asemănător au şi „bărbaţii XX”.
Sindromul Klinefelter 47, XXY
TESTUL CROMATINEI SEXUALE n n n n oferă informaţii privind numărul cromozomilor sexuali stabilirea sexului genetic (XX sau XY), a unor anomalii nr. cromozomi sexuali. test simplu, ieftin şi util în practică, în situaţii clinice bine definite. Diagnosticul final va fi pus însă numai prin analiza cromozomică. Testul cromatinei X se recomandă, în prezent, în două situaţii frecvente: n o anomalie a organelor genitale externe (hipospadias testiculi necoborâţi congenital, micropenis etc) sau o stare intersexuală evidentă; cromatina sexuală va permite stabilirea sexului genetic. n Semne clinice care ar putea evoca fenotipul unor disgenezii gonadice (în special 45, X sau 47, XXY): n la fete: n hipostatură neproporţională; n pubertate întârziată, caractere sexuale secundare reduse, n amenoree primară sau secundară precoce; n la băieţi: talie înaltă, pe seama membrelor inferioare; caractere sexuale secundare reduse; ginecomastie, testicule mici.
Corpuscul Barr – cromatina sexuală X Nr. Barr = X - 1
Testul F Cromatina sexuală Y: - reprezintă braţul q al cromoyomului Y, heterocromatină constitutivă, în cel. somatice 46, XY sau în spermatozoizii 23, Y; - se prezintă sub forma unui corpuscul F (fluorescent) de 0, 25 m în nucleele interfazice; - testul Feste util în identificarea crs Y (determinarea prenatală a sexului): NR crs. Y = NR corpusculilor F 46, XX – 0 46, XY – 1 47, XYY – 2 47, XXY - 1 48, XXYY – 2 46, X, i(Yp) – 0 46, X, i(Yq) – 1 (0, 5 m)
ABERAŢIILE CROMOZOMIALE = ANOMALIILE CROMOZOMIALE DE STRUCTURĂ n Cauze: n n n Acţiunea clastrogenă a unor agenţi chimici, fizici sau virusuri în timpul replicării ADN determină rupturi în ambele catene ale ADN urmate de reunirea anormală a fragmentelor cromozomice; Existenţa unor situsuri fragile pe anumiţi crs rupturi specifice; Conjugarea anormală a crs în timpul PI meiotice cu crossing-over inegal.
Anomalii cromozomiale constituţionale ECHILIBRATE (0, 43% - 1: 232 nn) n n Pot fi: n translocaţii reciproce n translocaţii robertsoniene n inversii Reprezintă rearanjamente cromozomiale Fenotipul purtătorilor poate fi normal sau patologic determinat de efectul poziţiei genelor pe crs. blocarea gametogenezei sau formarea de gameţi anormali Reprezintă cauza tulburărilor de reproducere: n Sterilitate n Avorturi spontane n nn morţi n nn plurimalformaţi.
Purtătorul de t 9/22 – MLC În regiunea H – protooncogena ABL n
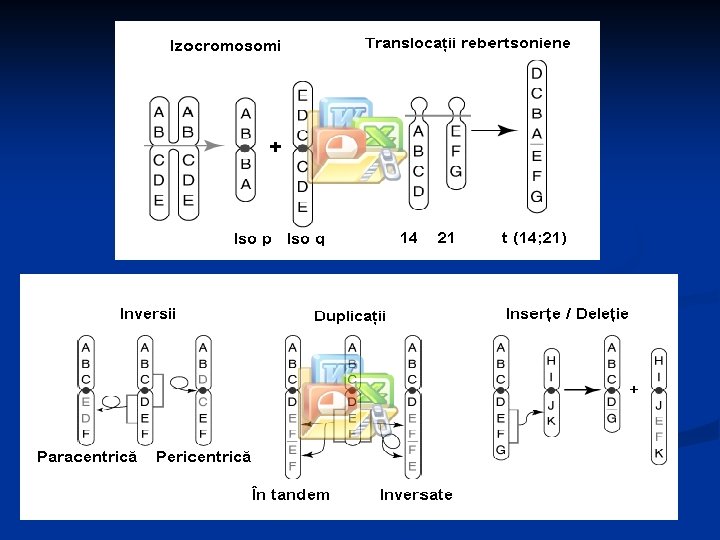
Mecanismele de producere ale aberaţiilor cromozomiale n Ruperea crs în unul sau două puncte urmată de: Pierderea fragmentului crs del, r; n Inversia fragmentului crs inv; n Inserţia pe alt crs – t rec şi t nerec; n Crossing-over inegal – del şi dup; n Ruperea a doi crs G sau D în regiunea centr şi unirea lor – rob; n Clivarea transversală a centromerului – ip sau iq. n
Deleţie (del) Terminală Interstişială
Inversie (inv) Paracentrică Pericentrică
Translocaţii (t) Reciprocă Cu inserţie Robertsoniană (rob)
Alte aberaţii cromozomale Crossing-over inegal – duplicaţie (dup) Cromozom inelar (r) Izocromozom (i, iso)
Consecinţele translocaţiei robertsonieine t(13 q 21 q)
Sindromul Anomalia crs Manifestări clinice majore Wolf Hirschorn 4 p- Prader – Willi del (15) (q 11 -q 13) crs patern Sindrom plurimalformativ congenital: microcefalie, hipotrofie staturo ponderală, dismorfie facială caracteristică, malformaţii cardiace grave, retard mintal sever. Hipotonie neonatală, dismorfie cranio facială caracteristică, obezitate, hipogonadism, retard mintal moderat, tulburări de comportament.
Sindromul Anomalia cromozomică Manifestări clinice majore Angelman del (15) (q 11 -q 13), crs matern Microcefalie, retard mintal sever, tulburări de mers şi echilibru, absenţa vorbirii, tulburări de comportament Williams del (7) (q 11. 23) Dismorfie facială caracteristică, stenoză aortică, laxitate articulară, hipostatură, retard mintal, dereglări psihice
Sindrom Anomalia crs. Manifestările principale Sdr. Langer – 8 q- 23 - q 24 Dismorfism cranio-facial, Gideon (trihoexostoze multiple, talie joasă, rino-falangeal urechi mari deformate, clinobrahidactilie, retard mental moderat. Sdr. Beckwit- 11 p+15 Widemanne Hernia funiculului ombelical, macroglosie, gigantism, hipoglicemie, microcefalie, malformaţii congenitale organelor interne.
Sindromul Cri du chat 46, XX, 5 p- sau 46, XY, 5 p-
Amprentare genică (imprinting) – inactivarea selectivă a alelei în dependenţă de originea ei parentală Aa Aa Aa
46, XY, 15 q- Sindrom Prader-Willy 46, XX, 15 q- Sindrom Angelman
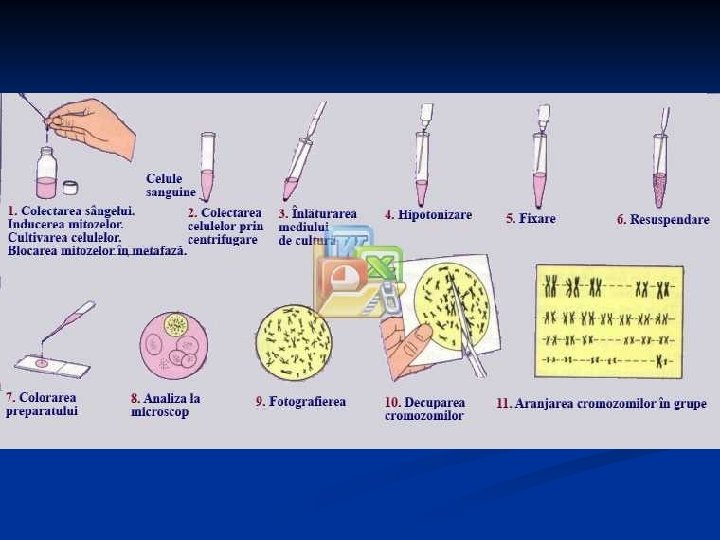
Studiul cariotipului uman Analiza crs metafazici colorare uniformă colorare în benzi G, Q, R, C, T Analiza crs prometafazici colorare în benzi G, R Analiza crs interfazici Testul cromatinei sexuale X şi Y Metode molecular citogenetice FISH, m. FISH SKY CGH *** NR benzi pentru setul haploid: 300 -400 m / 550 pm / 850 p
Tehnici de citogenetică – de analiză a cariotipului: Pentru identificarea anomaliilor de număr – analiza cromozomilor metafazici cu colorate omogenă. Pentru identificarea anomaliilor de număr şi de structură – analiza cromozomilor metafazici cu colorate diferenţiată Q, G, R; analiza cromozomilor prometafazici cu colorare diferenţiată Q, G, R. Pentru identificarea polimorfismului cromozomial – analiza cromozomilor metafazici cu colorate diferenţiată C, T.
INDICAŢIILE ANALIZEI CROMOZOMILOR UMANI n (1) Copiii cu anomalii congenitale multiple (minore/majore) asociate cu: n n (2) Debilităţi mintale (indiferent de grad) de cauze nedeterminate şi/sau tulburări de comportament – asociate cu: n n n tulburări de creştere prenatală, întârziere în dezvoltarea psiho-motorie postnatală, anamneza familială – tulburări reproducere. dismorfie facială, anamneză familială pozitivă – teste pentru X fragil). (3) Dacă în situaţiile (1), (2) se identifică o anomalie de structură neechilibrată (monosomie sau trisomie parţială) se va studia cariotipul. n părinţilor anomaliile cromozomială echilibrată; n rudelor gr. I (4) Stări intersexuale, pentru stabilirea sexului genetic (XX sau XY) sau anomalii ale cromozomilor sexuali. (5) Tulburări de dezvoltare pubertară semne de disgenezie gonadică: n spermogramă anormală (azo- sau oligospermie) n amenoree primară sau amenoree secundară precoce.
INDICAŢIILE ANALIZEI CROMOZOMILOR UMANI Cupluri cu tulburări de reproducere Hemopatiile maligne, Sindroame cu instabilitate cromozomială (sindromul Bloom, anemia Fanconi, sindromul Nijmegen, sindromul ICF ş. a). Depistarea efectului mutagen al expunerii profesionale sau accidentale la radiaţii ionizante şi unele substanţe chimice (clastogene). În DIAGNOSTICUL PRENATAL, studiul cromozomilor în celulele fetale este indicat la femeile gravide. n n n n n peste 35 de ani; părinţi purtători de mutaţii cromozomiale echilibrate; copil cu o anomalie cromozomială de novo (deşi cariotipul părinţilor este normal este posibil un mozaicism gonadic prenatal); semne ecografice de alarmă pentru stabilirea sexului genetic, în cazul mamelor purtătoare de mutaţii recesive gonosomale.