OPIOID ANALGESICS SBP 4301 BY ASSOC PROF DR

OPIOID ANALGESICS SBP 4301 BY ASSOC PROF DR ROSLIDA ABD HAMID 10 TH APRIL 2019

� Defined as agents that decrease pain � Categorized into a few classes: ◦ Non-steroidal anti-inflammatory drugs (NSAIDs) ◦ Anaesthetics ◦ CNS depressants ◦ Opioid analgesics ◦ Etc ØOpiod analgesics=narcotic analgesic=cause sleep or lose consciousness(narcosis) ØNarcotic associated with addictive properties misleading as not all opioids are addictive
=")
History � � � juice@latex from the unripe seed pods of poppy (Papaver somniferum)= opium The oldest medication used by human 3500 BC Surturner (1803) isolated an alkaloid from opium = morphine Codeine thebaine, papaverine etc Morphine undergo structure modification to Ethylmorphine (1898) and diacetylmorphine (heroin) in 1874 –nonaddicting analgesic, antidiarheal and anti-tussive

Opiate/opioid � Opiates-narcotic analgesic structurally related to morphine � Opioids : synthetic, semi-synthetic, naturally occurring, and endogenous compounds that interact with opioid receptors in the body. � Antagonized by an opioid antagonist eg naloxone � Opioid receptors=neuronal located proteins to which it binds and initiate biological response

Clinical Significance Opioid agonists and partial agonists act at δ, µ, ĸ receptors. They have subtypes which provide varying degrees of analgesia, euphoria/dysphoria, CNS depression, potential of tolerance. By modifying their structures, properties can be changed to develop agents that require more@less hepatic metabolism, thus affect the duration OA & bioavailability. Other changes in the chemical structure can yield agents with much higher affinity for analgesic receptors that correspond on mg to mg basis. Other alteration lead to improved profile regarding respiratory depression, emesis, tolerance and allergenicity. By altering the affinities for some receptors, the addictive properties may also be manipulated. � Selection of drugs to specific patients can be improvised through the understanding of relationship of chemical structure to biological activity �

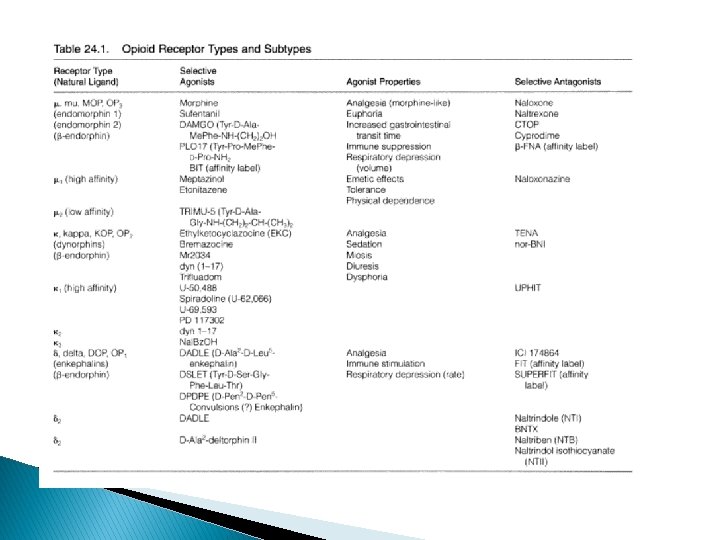
Characteristic features � Opioid peptides � Opioid receptors � Orphan opioid peptides � Endogenous receptors � Mu (µ) � Kappa (ĸ) � Delta (δ)
 Proenkephalin A — Met- and Leu-Enkephalin (ii) Propiomelanocortin")
Opioid peptides Precursor Opioid peptides (i) Proenkephalin A — Met- and Leu-Enkephalin (ii) Propiomelanocortin — β-Endorphin (PMOC) (iii) Proenkephalin B — Dynorphin, and α(Prodynorphin) Neoendorphin Exhibit analgesic activity at both supraspinal and spinal sites

Opioid Peptides n Greek, kephalē, which means “head” gives the name enkephalins. 2 enkephalins differ in the fifth amino acid one has a Methionine and the other a Leucine residue: Met–enkephalin and Leu–enkephalin n pro–enkephalin and few more endogenous peptides with opioid activity have been found, the endorphins and the dynorphins, n The opioid receptor has been subtyped into three distinct types based on these three types of endogenous peptides: (Endorphins –Mu, dynorphins – kappa, enkephalins – delta) n In all three types of peptides the amino terminus is always the same four amino acids (H 2 N–Tyr–Gly–Phe–)

 ; (DOR) � OP")
Opioid receptors � OP 1—Receptors : Delta opioid receptors (δ) ; (DOR) � OP 2—Receptors : Kappa opioid receptors (κ) ; (KOR) � OP 3—Receptors : mu opioid receptors ; (MOR) � Located either in human brain or spinal cord tissues � Mu and Kappa are of clinical use of analgesics � Receptor subtypes- receptor types that are coupled to different signal transduction systems
 � never got bound to the classical opioid peptide or")
Orphan receptor (OP 4) � never got bound to the classical opioid peptide or prevailing antagonists or known non-peptide agonists with high affinity � But a heptadecapeptide @ nociceptin@ orphanin as its endogenous peptide bound to the OP 4 � Resembles dynorphin A in structure (Phe replaced Tyr at N-terminus) � it causes hyperalgesia by releasing Substance P

Identification & activation of opioid receptors � Depends on the discovery of selected agonists/antagonists, identification of sensitive assay technique, cloning of rec. proteins � Most useful techniques: 1) Radioligand binding assay on brain tissue (2) electrically stimulated peripheral muscle preparations � Signal transduction mechanism for all receptors via Gi/o proteins � Activation of receptors linked through G proteins adenylate cyclase activity inhibited decrease in c. AMP K+ efflux Ca 2+ voltage gated channel closed hyperpolarization of nerve cells inhibited nerve firing
 � endomorphin-1 . (Tyr-Pro-Trp-Phe-NH 2) and endomorphin-2 (Tyr-Pro-Phe-NH 2). showed")
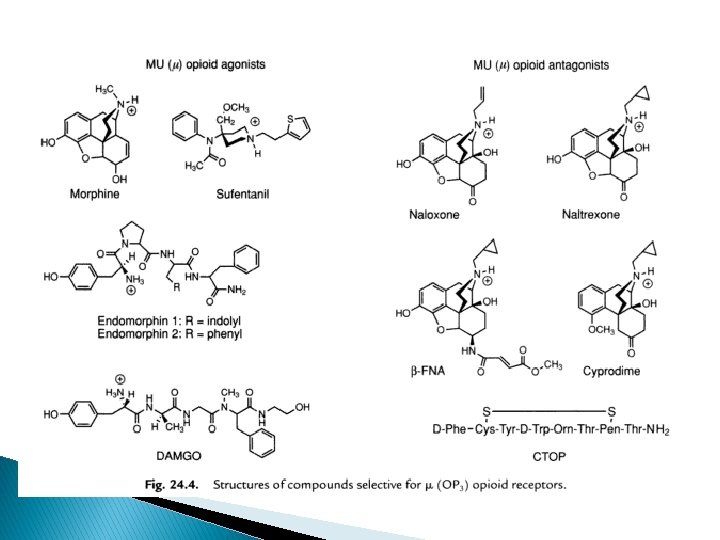
µ- opioid receptor (MOR) � endomorphin-1 . (Tyr-Pro-Trp-Phe-NH 2) and endomorphin-2 (Tyr-Pro-Phe-NH 2). showed an extremely high degree of selectivity for the mu (OP 3) receptors exclusively. � A plethora of therapeutically potent and useful compounds, such as : morphine, sufetanil, endomorphin-1, endomorphin-2, are potent Mu (μ) opioid agonists. � Naloxone, Cyprodime, Naltrexone are Mu (μ) opioid antagonists. � Practically all the ‘opioid alkaloids’ and most of their synthetic structural analogues are precisely the µ selective agonists.

� Three ‘drug substances’, namely : morphine, normorphine, and dihydromorphinone are found to have 10– 20 times more mu receptor selectivity. � Sufentanil, peptides DAMGO, demorphin 100 x selectivity � observed activities are : decreased gastric motility, emesis, tolerance, analgesia, respiratory depression and withdrawal symptoms.

� Cyprodime - most selective non-peptide µantagonist i. e. , 100 x than δ ; 30 x than ĸ � Naltrexone and Naloxone are recognized as opioid antagonists which exhibit only weak selectivity (5 to 10 fold) to MOR � µ 1 – mediate pain transmission eg naloxoeazine selective inhibitor to µ 1 receptor � µ 2 - control respiratory depression
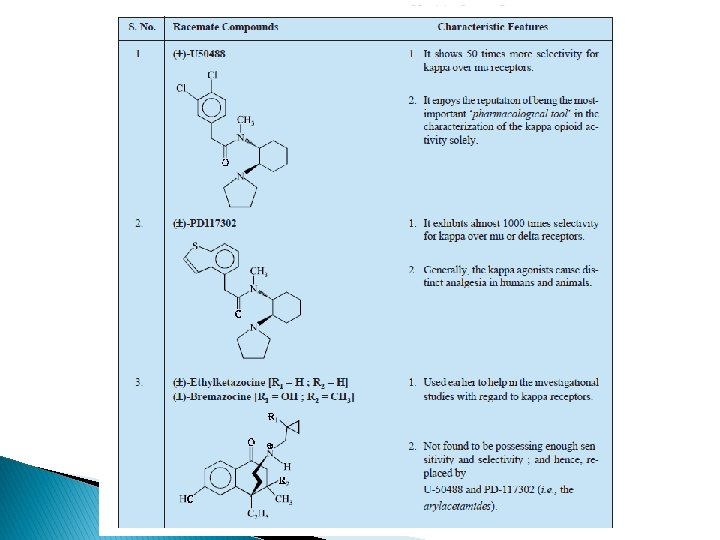
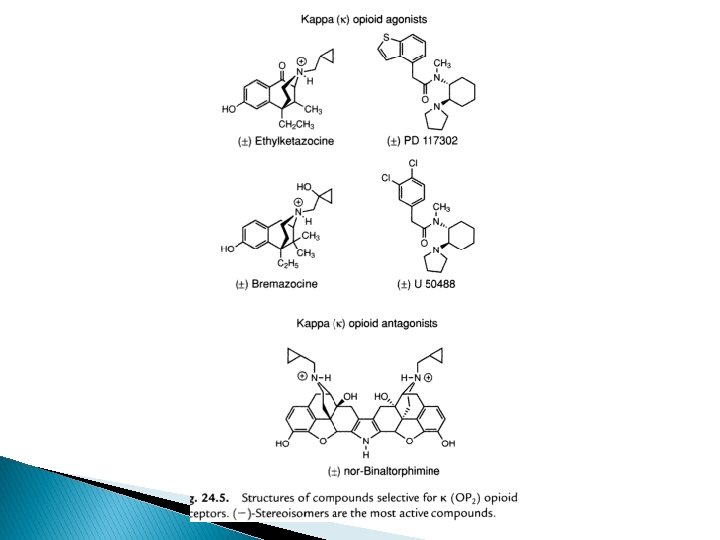
 receptor � Ethyl ketazocine & bremazocine (6, 7 -benzomorphan) selective to KOP")
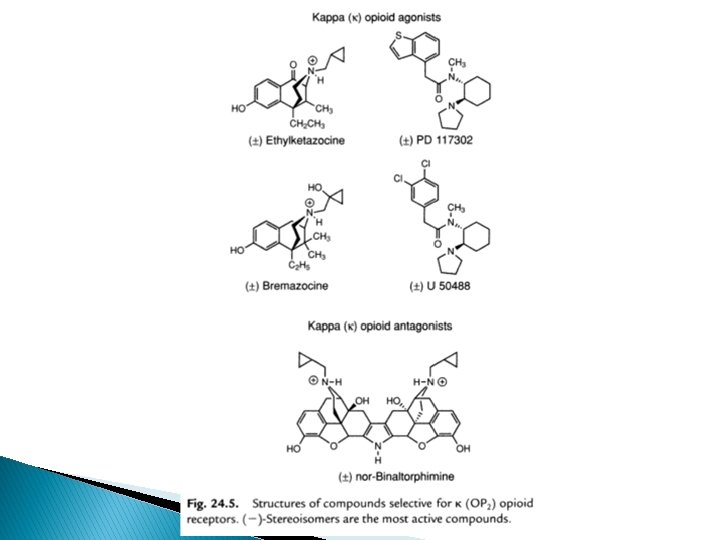
Kappa (ĸ) receptor � Ethyl ketazocine & bremazocine (6, 7 -benzomorphan) selective to KOP – diminished later � Arylcetamides binds to K subtypes receptor � ĸ agonists produce analgesia; sedation, diuresis, dysphoria; lack respiratory depression, constipating, strong addictive( euphoria, physical dependence) � Peptides related to dynorphin are natural agonists for KOR, selectivity compared to mu is not high � Norbinalthorphimine- K antagonist, selectivity 100 fold than DOR@MOR, but without any medical use
 receptors � Enkephalin only slightly selective to δ over µ � But")
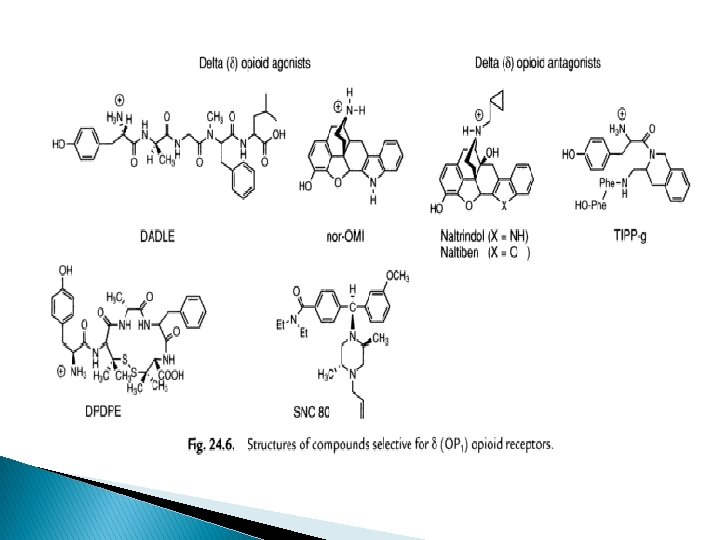
Delta (δ) receptors � Enkephalin only slightly selective to δ over µ � But changes in AA selection in enkephalin will increase the selectivity � Peptides most often used; [D-Ala 2, D-Leu 5]enkephalin (DADLE), [D-Ser 2, Leu 5] enkephalin-Thr (DSLET), cyclic pentide [D-Pen 2, D-Pen 5] enkephalin (DPDPE) � non-peptide agonist, morphindoles deriv, selectivity to DOR ie SNC 80 � Naltrindol, naltriben-selective non-peptide δ receptor antagonist � Peptidyl antagonist- TIPP, TIPP-Ψ � Antagonist as immunosuppressants and treating cocaine abuse
, minimize euphoric/aversive")
Rehabilitation of opioid addiction � Oral methadone mantains the addicted (tolerant state), minimize euphoric/aversive mood swings, attenuate drug craving � L-α-acetylmethadol, buprenorphine may replace methadone � Failure to alleviate drug-craving behavior of recovering addict, � Treated detoxified opioid addict with antagonists eg naltrexone block readdiction and curb addict’s craving urge � Naltrexone treatment in alcoholic

SAR of mu-agonist
, 6(S), 9(R),")
Morphine � Composed of 5 fused rings; 5 chiral centres , 5(R), 6(S), 9(R), 13(S), 14(R) � Isomer levo-[(-)] rotatory � Minor changes affinity and intrinsic activity of new compounds different physicochemical(solubility, partition, coefficient diff pharmacokinetic affect in vivo activity profile
 – necessary components in every potent")
�A ring & basic N (in ionized form) – necessary components in every potent µ-agonists � If either one exist alone- not sufficient for opioid activity; ; needs other pharmacophoric group � N – 30 amine-good opioid activity; size of Nsubstituent dictate its potency and whether it’s agonist@antagonist � Increasing 3 C 5 C= antagonist at all receptor types µ-agonist@antagonist

Summarizingthe the. Effectsofofalteringchainlengthon on. NNofof morphinestructure

Ideal opioid � Oral active drugs retain its analgesic morphine-like properties with reduced or no tolerance, physical dependence , respiratory depression, emesis, constipation � Focused on agents that act via other receptor types@ subtypes@nonopioid neurotrasmitter systems

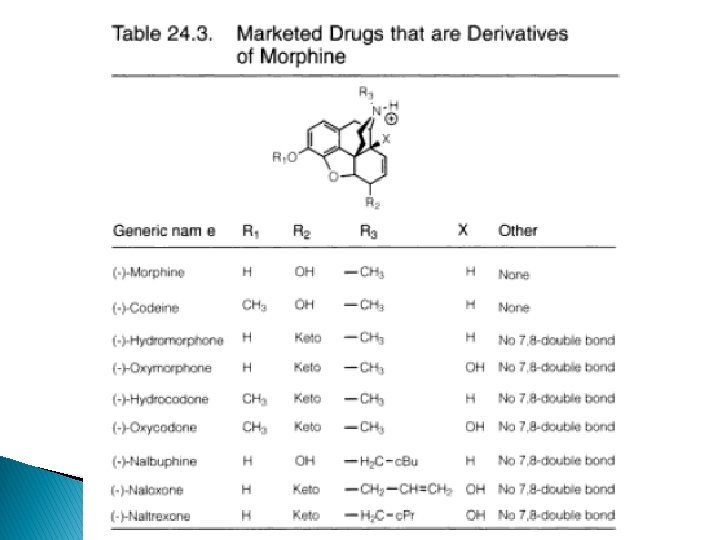
3 -phenolic hydroxy groups � Codeine- 3 methoxy derivative of morphine; weak mu -agonist; potent antitussive � Heroin- 3, 6 -diacetyl derivative; low affinity for mureceptor, high lipophilicity penetration to BBB hydrolyse 3 -acetyl group in the body 6 acetylmorphine � Produce euphoric rush drug abuse � Changes in C-ring increased activity; eg hydromorphone (7, 8 -dihydro-6 -keto-deriv), 8 -10 x more potent than morphine � Hydrocodone (3 -methoxy deriv) of hydromorphone is more active than codeine

Oxymorphone Nalbuphine heroin Hydromorphone Hydrocodone
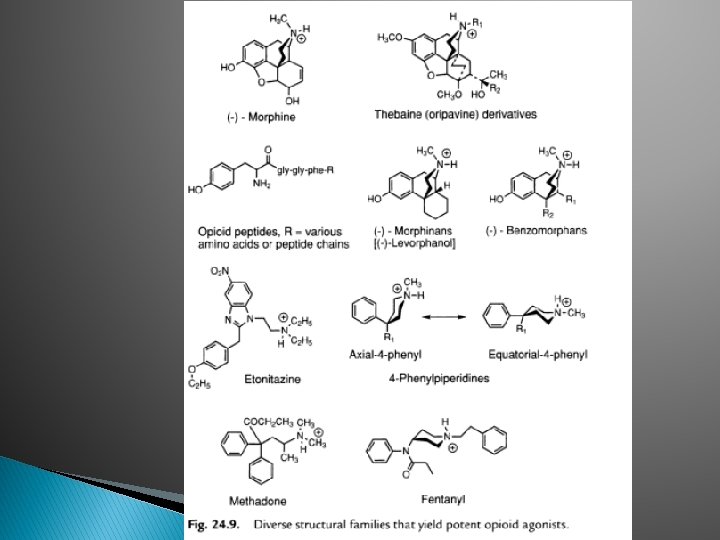

� Figures shown that morphine can be built up @broken down to yield compounds with potent agonist activity � Thebaines + dienophiles oripavines � Eg: buprenorphine (20— 30 x), etorphine (1000 x potent than morphine)

14α-keto-6 -hydroxy derivatives � Opium alkaloid, thebaine can be converted to 14α-keto-6 -hydroxy derivatives � 14α-OH group Increase mu-agonist activity, decrease antitussive activity � Oxycodone (3 -OCH 3 -N-CH 3 -)potent>morphine in parenteral � Oxymorphone (3 -OH-N-CH 3 -)- is 10 x potent than morphine � N-methyl substitute with N-cyclobutylmethyl, 6 keto 6α-OH of oxymorphone nalbuphine acts via K receptors; ½ potency analgesic of morphine ◦ Antagonist at µ receptor
 and N-cyclopropylmethyl (Naltrexone) noroxymorphone are “pure’ opioid antagonist - µ")
� N-allyl (Naloxone ) and N-cyclopropylmethyl (Naltrexone) noroxymorphone are “pure’ opioid antagonist - µ receptor selective, antagonist at all ORs � Etorphine and buprenorphine-Thebaine (oripavine) derivs

3, 4 -epoxide bridges and morphinans � Without 3, 4 -epoxide bridge=morphinan � Synthesis yields levo (-)isomers=opioid activity; dextro-antitussive � Levorpharnol (µ agonist) 8 x potent than morphine in analgesic � Butorpharnol ( µ-antagonist; ĸ agonist) Butorphanol Levorphanol Butorphanol

Benzomorphan � Minus epoxide ring and C-ring in morphine � Pentazocine- K agonist on analgesia; u antagonist and weak agonist at δ receptor � At higher dose, produce dysphoria, act at µ and δ receptor � Phenazocine (N-phenylethyl)- a u agonist 10 x potent than morphine � Aminotetralins-analogues of A & B rings – dezocine ( mixed agonist/antagonist) � � Bremazocine-200 x morphine Not addictive, no resp depression

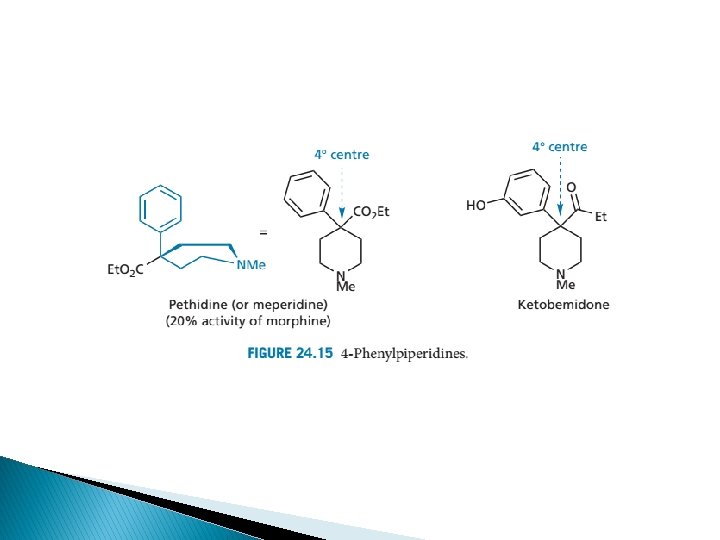
4 -phenylpiperidine �A and D-ring analogues-minus B, C and E rings � Meperidine – u agonist; ¼ potency of morphine � Useful in medical procedures due to short duration bcos esterases hydrolysis to zwitterionic metabolites � Reversed esters have higher potency meperidine

Anilidopiperidines � Structural modification of 4 phenylpiperidine 4 -anilidopiperidine = fentanyl-u agonists � Structural changes alfentanil, sufentanil � Produce analgesia at much lower dose than is necessary to cause resp. depression
-isomer produce analgesia")
Diphenylheptanone � Removing ring B, C, D, E � methadone � (-)-isomer produce analgesia � Reduction of keto, acetylation of OH acetylmethadols � Variations of methadone structures lead to discovery of antidiarheal opioids of loperamide and diphenoxylate

µ antagonists � 3 -phenolic group and N-allyl ie N-CBM, N-CPM replaced N-methyl � Compounds behaving u antagonist may act as agonist at other receptor types � Except buprenophine (N-CPM substituent)-; partial u agonist (partial u antagonist) � Pure antagonists ( act at all receptor types) are Naloxone ( N-allyl substituent) and naltrexone(NCPM substituent) derived from noroxymorphone � 14α-hydroxy groups important for pure antagonistic properties Buprenorphine

Noroxymorphone Naloxone Naltrexone

SAR of ĸ receptor agonists � Related to u antagonists =K agonists � Agonist activity enhanced if O group is placed at 8 -position (eg: ethylketazocine) or into Nsubstituent (eg: bremazocine)

SAR of δ receptor agonists � Least developed among the opioid compounds � Several selective δ agonists have potential convulsive actions thus withdrawn
, have Leu- or Met-enkephalin")
SAR of opioid peptides � All endogenous peptides (exc endomorphans), have Leu- or Met-enkephalin as their first five amino acid residues � Tyr as I 0 AA position is essential for activity. Removal of phenol hydroxy group at N will abolish activity. Tyr 1 free amino alkylated to give agonist@antagonist � Removal@changed of Enkephalin structure of Phe 4; x activity � Enkephalins have several low-energy conformations; bound at different receptor types/subtyeps � L-AA replaced by D-AA induce resistancy to several peptidase’s action thus rapidly degrade natural endorphins


� Conversion of terminal carboxyl group into amides@alcohol will protect the compound from carboxy peptidases � D/L-AA into enkephalins structure affects its conformational stability � Structural changes which highly restrict the conformational mobility of the peptides is useful for discovery of receptor-peptides selectivity

Enkephalin peptides � Lengthening carboxyl terminus-affinity to receptor type � Dynorphin; affinity to KOR � First four AA [Tyr-Gly-Phe]-essential for peptide ligands to bind to any receptor types � N-terminus AA-bring message to Receptor � + AA to C terminus address the msg to specific receptor

Metabolism of the opioids � Poor oral vs parenteral dose of morphine due to extensive 1 st pass metabolic conjugation of morphine at phenolic (3 -OH) position. � Glucurudination at 3 -OH undergoes enterohepatic cycling; increased oral dose � N-Demethylation Normorphine (decreased activity and bioavailability; undergoes N, Oconjugation excretion
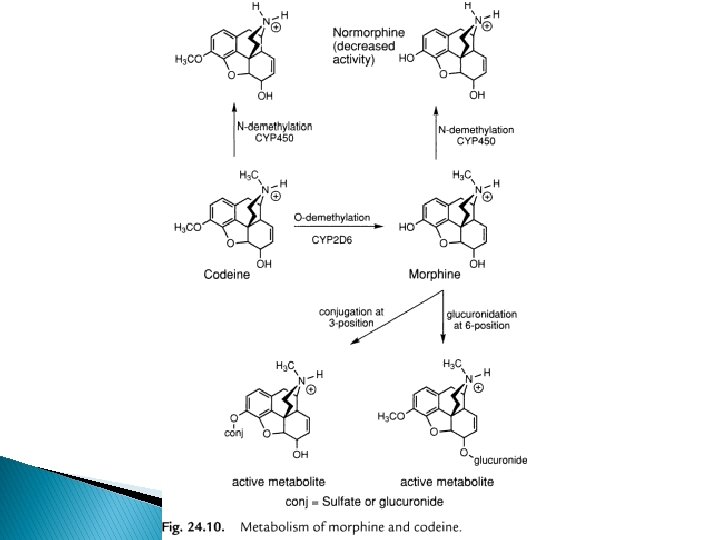

� 10% of oral dose of codeine is Odemethylated by CYP 2 D 6=morphine analgesia � Antitussive of codeine comes from the unmetabolized drug at non-opioid receptor, thus not affected by lack of CYP 2 D 6 � Compounds more lipophilic than morphine eg levorphanol have better oral activity � Compounds with N-alkyl larger than –CH 3 get N-dealkylated as major route of inactivation
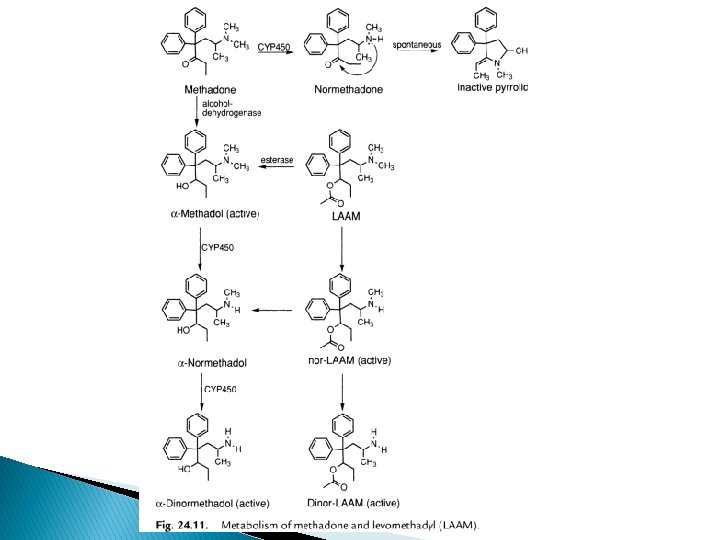

� Major route of inactivation of methadone via demethylation and cyclization of 2 o amine into inactive pyrrole deriv. � Keto group reduced by alcohol dehydrogenase methadol (less active than methadone) � N-demethylation produced normethadol and dinormethadol as active analgesics with increased t 1/2 � Metabolites have long duration action � LAAM (levo-α-acetylmethadol)longer acting, prodrug
-morphine sulfate � 3 - 6 x potent when")
Specific drugs �µ agonist ◦ (-)-morphine sulfate � 3 - 6 x potent when given i. m than oral �Due to extensive first pass 3 -0 glucurodination of morphine (inactive metabolite) �T 1/2 i. m dose (10 mg/kg) ~ 3 hrs �Reduced dose for renal failure, geriatric, pediatric patients �By oral (60 mg); maintenance dose 20 -30 mg every 4 hours
-Codeine phosphate ◦ Weak µ agonist; 10% oral dose metabolized into morphine ◦")
� (-)-Codeine phosphate ◦ Weak µ agonist; 10% oral dose metabolized into morphine ◦ Parenteral route is not recommended as it will cause the release of histamine sufficient for hypertension, pruritus and allergic responses � Hydromorphone hydrochloride (Dilaudid) ◦ Potent µ agonist; 8 x than morphine ◦ Addicting properties
; potent µ agonist; 1, 4 -OH has")
� � � � Oxymorphone hydrochloride (Numorphan); potent µ agonist; 1, 4 -OH has low antitussive properties (-)-Levorphanol Bitartrate (Levo-dromoran); 6 X > morphine ; potent µ agonist; longer duration (4 -6 hrs), t 1/2 clearance=11. 4 hrs Hydrocodone bitartrate : moderate pain, combination with acetaminophen; oral (-)-Oxycodone hydrochloride; equipotent to morphine; 3 -OCH 3 caused lower oral: parenteral dose ratio Meperidine hydrochloride (Demerol); µ agonist 1/10 than morphine on im. Strong adverse reaction with MAO inhibitors, less constipation; not inhibit cough (+)-Tramadol HCl; Analgesic activity from (+)-isomer and neurotrasmitter reuptake blocking effect of (-)-isomer. reduced effect for those who lack CYP 2 D 6@ taking CYP 2 D 6 inhibitor. Non addicting (+)- Methadone hydrochloride; all activity caused by (-); greater oral potency and longer duration. Used for ca pts; good drug for maintenance of addiction but not ideal Propoxyphene hydrochloride ; weak µ agonist; mixtures with NSAIDs. oral dosage; dose approx. approach analgesic efficacy of morphine is toxic

Meperidine HCl Propoxyphene HCl Tramadol HCl Methadone HCl
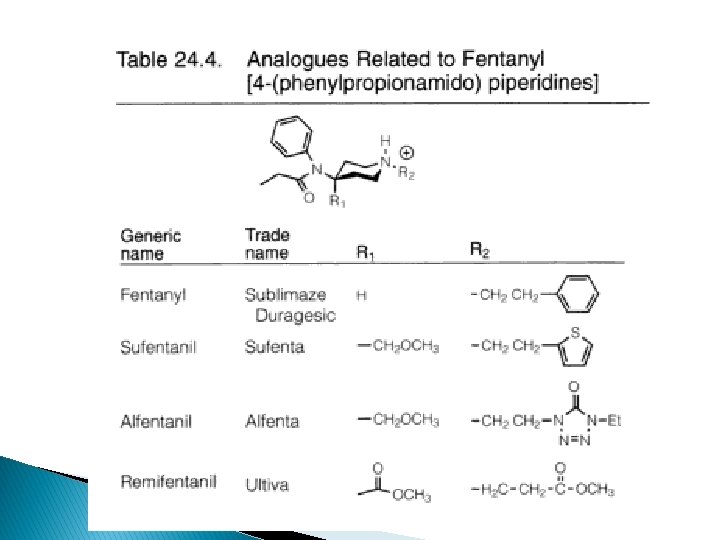

� Fentanyl; µ agonist, 80 x potent than morphine ◦ Combination w NO and dropiredol for anaesthesia ◦ Shorter duration (1 -2 h), no histamine release with iv � Sufentanil citrate (Sufenta); increase activity via SAR; 600 -800 x potent; shorter duration than fentanyl. Via iv � Alfentanil HCl (Alfenta): changes of thiophene ring decrease potency (-25 x). Penetrate BBB, thus shorter duration, fast OA, iv � Remifentanil HCl (Ultiva); 15 -20 x than alfentanil; short duration due to ester group rapidly hydrolyzed to inactive-COOH by serum and tissue esterases independent of liver/renal function of pts
-buprenorphine HCl (Buprenex); potent partial agonist at MOR, KOR,")
Mixed agonist/antagonist � � � (-)-buprenorphine HCl (Buprenex); potent partial agonist at MOR, KOR, antagonist at DOR; incapable of producing tolerance & addiction comparable to full µ agonists. 20 -50 X >morphine ◦ Maximal analgesic, less resp. depression ◦ Block high dose heroine ◦ Treating opioid dependence ◦ Naloxone not effective antagonist to buprenorphine (-)-Butorphanol tartrate (Stadol); strong agonist at KOR (5 x potent than morphine), antagonist at MOR (1/6 of naloxone); . Low abuse potential. First pass metabolism Nalbuphine HCl (Nubain); antagonist at MOR, agonist at KOR. ¼ potent than naloxone; side effect=K agonists Pentazocine HCl; weak antagonist at MOR (1/30 of Naloxone), agonist at KOR. Side effects=K agonists. Major abuse; + antihistamine, tripelenamine increase euphoric, decrease dysphoric. Dezocine (Dulgan); primary amine, =buprenorphine; partial agonist at DOR and MOR, little effect at KOR. High abuse potential. Equipotent to morphine. For postoperative pain and cancer-induced pain
-Buprenorphine HCl (-)-Pentazocine HCl (-)-Butorphanol Tartrate Dezocine Nalbuphine")
(-)-Buprenorphine HCl (-)-Pentazocine HCl (-)-Butorphanol Tartrate Dezocine Nalbuphine

Opioids use anti-diarrheal � SAR of 4 -phenylpiperidines antidiarrheal � Agonists at MOR and DOR have strong inhibition on the peristaltic reflex on intestine � Occurs due to intestinal wall innervation by opioid agonists synapse into cholinergic neurons inhibit ACh release inhibit peristalsis � Any µ agonists used cause constipation as side effect. Not used as antidiarrheal due to abuse and addiction potential � Synthetic agent that are structural combination of meperidine and methadone are extensively used as antidiarrheal
 � Difenoxin HCl with Atropine sulfate (Motofen)")
� Diphenoxylate HCl with Atropine sulfate (Lomotil) � Difenoxin HCl with Atropine sulfate (Motofen) � Loperamide HCl (Imodium) Loperamide HCl Diphenoxyl HCl + Atropine Sulfate

� Combination w atropine enhance the block of ACh-stimulated peristalsis. Adv effect of atropine eliminate the abuse potency of opioid. Diphenoxylate alone has low µ agonist. Metabolites (difenoxin) 5 x more potent. High dose of diphenoxylate will cause euphoria and addiction.

Enkephalinase inhibitor � Acetorphan pro drug of Thiorphan � Orally dosed acetorphan caused antidiarrheal by inhibition of intestinal secretion � Complementary effect when combined with loperamide by decreasing GI transit time
 isomers= (-) -isomer �")
Opioid agents as antitussive � Rigid structured opioids � (+) isomers= (-) -isomer � 3 -OCH 3 deriv. of morphine (Codeine & hydrocodone), � better oral activity, decrease abuse potential of OCH 3 derivatives make them preferred as antitussive � 14α-OH (oxycodone) reduces the antitussive ◦ Codeine, less abuse potential ◦ Hydrocodone bitartrate; 3 x >codeine, analgesic, abuse potential ◦ Dextromorphan HBr; (+)-isomer of 3 -OCH 3 form of levorphanol; not an opioid, retain centrally acting antitussive; less than codeine effectiveness. In non prescription cough formulaion

- Slides: 71