ONCOGENES AND CANCER MCB 720 Susan Evans John

ONCOGENES AND CANCER MCB 720 Susan Evans John Kopchick

ONCOGENES AND CANCER MCB 720 1/07

• Statistics • Introduction to cancer and oncogenes • Compare tumor suppressors and oncogenes • Tumor progression • Mechanisms of oncogenes • Examples of mutations in oncogenes

Mortality for Leading Causes of Death, United States-2001 Number of deaths Percent of total Heart disease 700, 142 29. 0 Cancer 553, 768 22. 9 Cerebrovascular disease 163, 538 6. 8 Lung disease 123, 013 5. 1 Accidents 101, 537 4, 2 Diabetes mellitus 71, 372 3. 0 Pneumonia and influenza 62, 034 2. 6 Alzheimer’s disease 53, 852 2. 2 Nephritis 39, 480 1. 6 Septicemia 32, 238 1. 3 Cause of death Source: US Mortality Public Use Data Tape 2001 National Center for Health Statistics Centers for Disease Control and Prevention, 2003

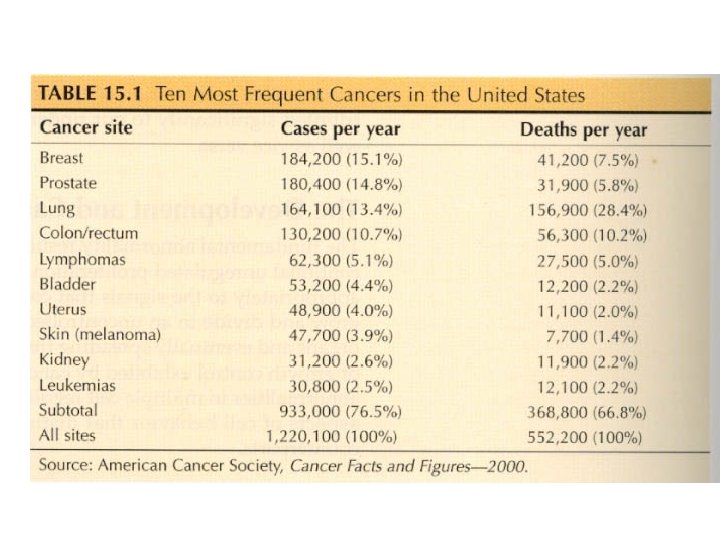
Who gets cancer? • • Over 1 million people a year 1 out of 2 men 1 out of 3 women ~80% of cancers occur in people over 55

Cancer

2004 Estimated US Cancer Causes Female Male Prostate Lung Colon Bladder 33% 11% 6% Breast Lung 32% 12% Colon Uterus 10% 6% Melanoma Non-Hodgkin Lymphoma Kidney Oral cavity Leukemia Pancreas All other 4% 4% Ovary Non-Hodgkin Lymphoma Melanoma Thyroid Pancreas Bladder All other 4% 4% 3% 3% 3% 2% 18% 4% 3% 2% 2% 20%

2004 Estimated US Cancer Deaths Female Male Lung Prostate Colon Pancreas 32% 10% 5% Lung Breast Colon Ovary 32% 10% 5% Leukemia Non-Hodgkin Lymphoma Esophagus Liver Bladder Kidney All other 5% 4% Pancreas Leukemia Non-Hodgkin Lymphoma Uterus Multiple myeloma Brain All other 5% 4% 4% 4% 3% 3% 3% 21%

Comparison between men and women of different ethnicities

Introduction to cancer
 (oncogenes) overactive inactive CANCER")
Cellular homeostasis Proliferation Arrest Survival Apoptosis Undifferentiated Differentiated (tumor suppressors) (oncogenes) overactive inactive CANCER

Definitions • Oncogene – a gene that when mutated or expressed at abnormally high levels contributes to converting a normal cell into a cancer cell • Proto-oncogene – the “normal” cellular progenitors of oncogenes that function to promote the normal growth and division of cells
 that")
Proto-oncogene to oncogene • An alteration occurs in a normal cellular gene (proto-oncogene) that makes the protein hyper-functional (oncogene) • Proteins involved in the cell signaling pathways are products of proto-oncogenes – Proliferative – Anti-apoptotic (survival) – Angiogenic

Tumor suppressors • Normally function to suppress the formation of cancer – Growth arrest – Apoptosis – DNA repair – Differentiation – Anti-angiogenesis

Tumor suppressors are recessive – require mutation of both alleles Oncogenes are dominant – mutation of 1 allele is sufficient
 1 st mutation (leads to accelerated cell division)")
Oncogenes Normal genes (regulate cell growth) 1 st mutation (leads to accelerated cell division) 1 mutation is sufficient for a role in cancer development.
 1 st mutation (susceptible carrier) 2 nd")
Tumor Suppressor Genes Normal genes (prevent cancer) 1 st mutation (susceptible carrier) 2 nd mutation or loss (leads to cancer) 2 mutations are necessary for a role in cancer development.

Comparison of Proto-oncogenes and tumor suppressors Property Tumor suppressor genes Proto-oncogenes Alleles mutated in cancer Both alleles One allele Germ line transmission of mutant allele frequent Rare (1 example) Somatic mutations yes Function of mutant allele Loss of function (recessive allele) Gain of function (dominant allele) Effects on cell growth Inhibit cell growth Promote cell growth

Normal cells • • • Anchorage dependence Growth factor dependent Contact inhibition Cytoskeletal organization Monolayer

Transformed cell • • • Unregulated growth properties Serum independence Anchorage independent No contact inhibition (form foci) May induce tumors in vivo

Tumorigenesis is a Multistep Pathway • Mutation of proto-oncogenes and tumor suppressor genes • Special combination -Yes • Particular order -Yes

Evidence for multistep cancer pathogenesis

Oncogene Cooperation myc ras Myc + ras

Mechanisms of collaboration • Multiple mutated genes disrupt multiple control points of anti-cancer mechanism • Synergistic/complementary activities • Cell tries to apoptose but selects for more aggressive cell with increased proliferative abilities

Multistep tumorigenesis • Initiation – 1 st mutation – Increased proliferation of a single cell • Progression – Additional mutations – Selection for more aggressive cells Clonal selection!

Initiation Progression Aggressive, rapidly growing tumor

With increase in histopathological abnormalities, there is an increase in the number of mutations at defined genetic loci

What causes the mutations that lead to cancer? • Anything that damages DNA – Physical agents (radiation) – Chemical agents (carcinogens) • Anything that stimulates the rate of mitosis – Viruses – Oncogenes – Tumor suppressor genes

How does damage affect function? • Increased and sustained activity on a gene or its protein product – Altered gene expression – Change in protein structure • Change in the specificity or function of the protein – Substrate specificity – Transactivation of different genes

Oncogenes

Activated oncogenes from DNA transfection Human bladder tumor cell line Isolate human DNA Alu probe (Alu PCR) Isolate DNA Result: A single human gene is responsible for transforming capability transfect 3 T 3 cells Transformed cells sequence Result: Human Ras Isolate DNA >99% mouse Compare to normal gene Result: Ras activation is due to a single point mutation

Oncogenes by location Secreted c-sis wnt 1 int 1 c-erb. B neu kit mas gsp gip Membrane associated ras src Cytoplasmic Nuclear myc myb fos jun rel erb. A abl fps raf mos Vav AKT Transmembrane

Oncogenes by function • • • Growth factors Growth factor receptors G proteins Intracellular kinases Transcription factors

Oncogenic mutations • GF receptors and signaling proteins can exist in active and inactive state • Active state is rapidly turned over – Dephosphorylation of kinases – Hydrolysis of GTP to GDP – Protein degradation • Oncogenic mutation alters protein product locked in the active state • Interpreted by cell as a continuous and unrestricted growth inducing signal

Mechanisms of conversion • • Insertional mutagenesis Translocation and inversion Amplification Point mutations

Chromosomal translocation

Myc genes Myc Max Basic HLH leucine Binds DNA Dimerization Myc-Max Increases trx Max-Max • Increased myc synthesis drives Max to partner with Myc • Myc has short half life • Max is stable Mad-Max Represses trx

Oncogenic mutation of myc 1 2 3 Proto-oncogene Translocation to Ig locus 2 Ig promoter and enhancer Oncogene Transcription Splicing m. RNA 2 3 Translation 3 • Chromosome translocation puts myc under control of strong promoter and enhancer • Increases the concentration of Myc. Max heterodimers thus increasing cell proliferation • Burkitt’s lymphoma Increased expression of normal myc protein

Translocation resulting in fusion of 2 genes Alters structure of normal c-abl protein

Cytoplasmic tyrosine kinase • C-abl encodes a cytoplasmic tyrosine kinase • Bcr promotes oligomerization • Bcr-abl fusion promotes activation of abl by oligomerization induced autophosphorylation • Philadelphia chromosome – translocation of chr 9 and 22 bcr kinase DBl-H abl SH 3 P 210 kinase bcr-abl P 185 kinase bcr-abl SH 2 Dbl-H SH 3 P Rho-GAP Kinase Tail P SH 2 SH 3 Kinase SH 2 Tail Kinase Tail

Signaling by secreted molecules Endocrine Y Y Paracrine Autocrine Y

Growth factor expression • Controlled at the level of gene expression – Autocrine • Cell produces a growth factor to which it also responds • Sis – encodes a variant form of PDGF – Astrocytomas – Increases cell growth – Paracrine • VEGF • Increases growth of endothelial cells • Secreted by tumor

Receptor activation

Rearrangement N terminal domain is replaced by a transcription factor that can associate with itself

Amplification Too much protein

Amplification Increased density induces dimer formation, autophosphorylation -thus constitutively active

Point mutation Constitutively active

Ras proteins • Activation of PTK receptor by ligand binding • Receptor associates with adaptor protein (grb 2) • Grb 2 SH 3 domain binds guanine exchange factor (Sos) • Sos activates Ras • Activated Ras interacts with protein kinase (Raf)

Regulation of Ras Inactive state Ras GTP GDP P GEF GTPase GDP GAP • Cycle is unidirectional • GTP bound is active form • Regulated by 2 classes of proteins • + regulator • -regulator Ras GTP Active state
 •")
Oncogenic mutations that constitutively activate Ras • Constitutive activation of GEFs (positive regulator) • Reduction of GAP activity (negative regulator) • Mutation of Ras gene – Cannot hydrolyze GTP

m. Sos 1 m. Sos 2 Ras-GRF INACTIVE GEF Ras-GDP ACTIVE Ras-GTP Interaction with target proteins Mutant Ras Constitutively active GAP P 120 gap Neurofibromin Gap 1 m X

Point mutation of PTK Induces dimerization in absence of ligand

Hormone Adenylyl cyclase Receptor bg a G protein a a Activated G protein ATP • G protein associated with inner surface of PM • Hormone bound receptor interacts with G protein • Stimulates release of GDP and exchange for GTP • Ga dissociates from complex • Stimulates production of c. AMP by adenylyl cyclase • c. AMP is a second messenger c. AMP

Regulation of G proteins Inactive state bg GTP hydrolysis bg a a GDP Hormone binding induces interaction of receptor with G protein GTP GDP GTP Target proteins Stimulates the exchange of GTP for GDP bg a GDP

Oncogenic mutations • Locking a subunit in an active state • Pituitary tumors – Gsp – encodes a mutated a subunit that blocks GTPase activity – Constitutive production of c. AMP • Thyroid tumors – Thyroid receptor mutated – Constitutive production of c. AMP

Therapeutic implications • High doses of retinoic acid can induce differentiation • Block growth factor/receptor interaction with antagonist • Tyrosine kinase inhibitors • Block protein interactions in signaling cascade (SH 2 domain) • Block membrane localization of Ras with farnesylation inhibitors

Mortality for Leading Causes of Death, United States - 1978 Number of deaths Percent of total Heart disease 729510 37. 8 Cancer 396992 20. 6 Cerebrovascular disease 175629 9. 1 Accidents 105561 5. 5 Pneumonia and influenza 58319 3. 0 Lung disease 50488 2. 6 Diabetes mellitus 33841 1. 8 Cirrhosis of liver 30066 1. 6 Arteriosclerosis 28940 1. 5 Suicide 27294 1. 4 Disease of infancy 22033 1. 1 Homicide 20432 1. 1 All others 248543 12. 9 Cause of death Source: Vital statistics of the United States, 1978 Modified from: CA – A Journal for Clinicians, Vol 33, 1983.

Who gets cancer? • • Over 1 million people a year 1 out of 2 men 1 out of 3 women ~80% of cancers occur in people over 55
 (oncogenes) CANCER")
Cellular homeostasis Proliferation Arrest Survival Apoptosis Undifferentiated Differentiated (tumor suppressors) (oncogenes) CANCER

Disruption of homeostasis Activation of oncogenes Inhibition of tumor suppressors

Definitions • Oncogene – a gene that when mutated or expressed at abnormally high levels contributes to converting a normal cell into a cancer cell • Proto-oncogene – the “normal” cellular progenitors of oncogenes that function to promote the normal growth and division of cells
 that")
Proto-oncogene to oncogene • An alteration occurs in a normal cellular gene (proto-oncogene) that makes the protein hyper-functional (oncogene) • Proteins involved in the cell signaling pathways are products of proto-oncogenes – Proliferative – Anti-apoptotic – Angiogenic

Tumor suppressors • Normally function to suppress the formation of cancer – Growth arrest – Apoptosis – DNA repair – Differentiation – Anti-angiogenesis

Tumor suppressors are recessive – require mutation of both alleles Oncogenes are dominant – mutation of 1 allele is sufficient
 1 st mutation (leads to accelerated cell division)")
Oncogenes Normal genes (regulate cell growth) 1 st mutation (leads to accelerated cell division) 1 mutation is sufficient for a role in cancer development.
 1 st mutation (susceptible carrier) 2 nd")
Tumor Suppressor Genes Normal genes (prevent cancer) 1 st mutation (susceptible carrier) 2 nd mutation or loss (leads to cancer) 2 mutations are necessary for a role in cancer development.

Comparison of Proto-oncogenes and tumor suppressors Property Tumor suppressor genes Proto-oncogenes Alleles mutated in cancer Both alleles One allele Germ line transmission of mutant allele frequent Rare (1 example) Somatic mutations yes Function of mutant allele Loss of function (recessive allele) Gain of function (dominant allele) Effects on cell growth Inhibit cell growth Promote cell growth

Definitions • Tumor – abnormal growth – Benign – Remain localized to tissue where they originated • Cancer – malignant – Invades surrounding tissue – Has the ability to metastasize

extravasation neovascularization Adherence Invasion Arrest in organ Angiogenesis and proliferation Establishment of microenvironment metastases Transport Primary neoplasm

Angiogenesis Definition: Process where tumor cells encourage the ingrowth of capillaries and vessels from adjacent normal tissue Tumor releases angiogenic factors Endothelial cells proliferate Tumor grows

How does a normal cell become a tumor cell? • Immortalization – Normal cells have fixed amount of doublings (~50) – Senescence • Telomerase activity decreases • Shorter telomeres – Crisis • Increase telomerase • Transformation

Immortalized/established cell line • • • Anchorage dependence Growth factor dependent Contact inhibition Cytoskeletal organization Monolayer

Transformed cell • • • Unregulated growth properties Serum independence Anchorage independent No contact inhibition (form foci) May induce tumors in vivo

Focus Forming Assay Lacks contact inhibition


Tumorigenesis is a Multistep Pathway • Mutation of proto-oncogenes and tumor suppressor genes • Special combination - yes • Particular order - yes

Evidence for multistep cancer pathogenesis

Cooperation between genes • Myc – few mice with tumors • Ras – more mice with tumors • myc + ras – all mice with tumors Conclusion: Complementary activities of 2 distinct oncogenes function collaboratively to create fully tumorigenic cells

Oncogene Cooperation myc ras Myc + ras

Mechanisms of collaboration • Multiple mutated genes disrupt multiple control points of anti-cancer mechanism • Synergistic/complementary activities • Cell tries to apoptose but selects for more aggressive cell with increased proliferative abilities

Multistep tumorigenesis • Initiation – 1 st mutation – Increased proliferation of a single cell • Progression – Additional mutations – Selection for more aggressive cells Clonal selection!


Initiation Progression Aggressive, rapidly growing tumor

With increase in histopathological abnormalities, there is an increase in the number of mutations at defined genetic loci

What causes the mutations that lead to cancer? • Anything that damages DNA – Physical agents (radiation) – Chemical agents (carcinogens) • Anything that stimulates the rate of mitosis – Viruses – Oncogenes – Tumor suppressor genes

How does damage affect function? • Quantitative model- increased and sustained activity on a gene or its protein product – Altered gene expression – Change in protein structure • Qualitative model- change in the specificity or function of the protein – Substrate specificity – Transactivation of different genes

Activated oncogenes from DNA transfection Human bladder tumor cell line Isolate human DNA Alu probe (Alu PCR) Isolate DNA Result: A single human gene is responsible for transforming capability transfect 3 T 3 cells Transformed cells sequence Result: Human Ras Isolate DNA >99% mouse Compare to normal gene Result: Ras activation is due to a single point mutation

Oncogenes by location Secreted c-sis wnt 1 int 1 c-erb. B neu kit mas gsp gip Membrane associated ras src Cytoplasmic Nuclear myc myb fos jun rel erb. A abl fps raf mos Vav AKT Transmembrane

Oncogenes by function • • • Growth factors Growth factor receptors G proteins Intracellular kinases Transcription factors

Oncogenic mutations • GF receptors and signaling proteins can exist in active and inactive state • Active state is rapidly turned over – Dephosphorylation of kinases – Hydrolysis of GTP to GDP – Protein degradation • Oncogenic mutation alters protein product so that it is locked in the active state • Interpreted by cell as a continuous and unrestricted growth inducing signal

Mechanisms of conversion • • Induced by virus Transduction Insertional mutagenesis May or may not be virus induced Chromosomal translocation and inversion Amplification Point mutations

Chromosomal translocation

Myc genes Myc Max Basic HLH leucine Binds DNA Dimerization Myc-Max Increases trx Max-Max • Increased myc synthesis drives Max to partner with Myc • Myc has short half life • Max is stable Mad-Max Represses trx

Oncogenic mutation of myc 1 2 3 Proto-oncogene Translocation to Ig locus 2 Ig promoter and enhancer Oncogene Transcription Splicing m. RNA 2 3 Translation 3 • Chromosome translocation puts myc under control of strong promoter and enhancer • Increases the concentration of Myc. Max heterodimers thus increasing cell proliferation • Burkitt’s lymphoma Increased expression of normal myc protein

Translocation resulting in fusion of 2 genes Alters structure of normal c-abl protein

Cytoplasmic tyrosine kinase • C-abl encodes a cytoplasmic tyrosine kinase • Bcr promotes oligomerization • Bcr-abl fusion promotes activation of abl by oligomerization induced autophosphorylation • Philadelphia chromosome – translocation of chr 9 and 22 bcr kinase DBl-H abl SH 3 P 210 kinase bcr-abl P 185 kinase bcr-abl SH 2 Dbl-H SH 3 P Rho-GAP Kinase Tail P SH 2 SH 3 Kinase SH 2 Tail Kinase Tail

Signaling by secreted molecules Endocrine Y Y Paracrine Autocrine Y

Growth factor expression • Controlled at the level of gene expression – Autocrine • Cell produces a growth factor to which it also responds • Sis – encodes a variant form of PDGF – Astrocytomas – Increases cell growth – Paracrine • VEGF • Increases growth of endothelial cells • Secreted by tumor

Receptor activation

Rearrangement N terminal domain is replaced by a transcription factor that can associate with itself

Amplification Too much protein

Amplification Increased density induces dimer formation, autophosphorylation thus constitutively active

Growth factor receptor EGF TGFb EGFR TGFR p 27 cyclin. D 1/cdk 4 Rb-E 2 F 1 Cyclin. D 1 – amplification associated with cancer cyclin. E/cdk 2 P Rb + E 2 F 1 S phase genes proliferation

Point mutation Constitutively active

Ras proteins • Activation of PTK receptor by ligand binding • Receptor associates with adaptor protein (grb 2) • Grb 2 SH 3 domain binds guanine exchange factor (Sos) • Sos activates Ras • Activated Ras interacts with protein kinase (Raf)

Regulation of Ras Inactive state Ras GTP GDP P GEF GTPase GDP GAP • Cycle is unidirectional • GTP bound is active form • Regulated by 2 classes of proteins • + regulator • -regulator Ras GTP Active state

m. Sos 1 m. Sos 2 Ras-GRF INACTIVE GEF Ras-GDP ACTIVE Ras-GTP Interaction with target proteins Mutant Ras Constitutively active GAP P 120 gap Neurofibromin Gap 1 m X
 •")
Oncogenic mutations that constitutively activate Ras • Constitutive activation of GEFs (positive regulator) • Reduction of GAP activity (negative regulator) • Mutation of Ras gene – Cannot hydrolyze GTP

Point mutation of PTK Induces dimerization in absence of ligand

Hormone Adenylyl cyclase Receptor bg a G protein a a Activated G protein ATP • G protein associated with inner surface of PM • Hormone bound receptor interacts with G protein • Stimulates release of GDP and exchange for GTP • Ga dissociates from complex • Stimulates production of c. AMP by adenylyl cyclase • c. AMP is a second messenger c. AMP

Regulation of G proteins Inactive state bg GTP hydrolysis bg a a GDP Hormone binding induces interaction of receptor with G protein GTP GDP GTP Target proteins Stimulates the exchange of GTP for GDP bg a GDP

Oncogenic mutations • Locking a subunit in an active state • Pituitary tumors – Gsp – encodes a mutated a subunit that blocks GTPase activity – Constitutive production of c. AMP • Thyroid tumors – Thyroid receptor mutated – Constitutive production of c. AMP

Therapeutic implications • High doses of retinoic acid can induce differentiation • Block growth factor/receptor interaction with antagonist • Tyrosine kinase inhibitors • Block protein interactions in signaling cascade (SH 2 domain) • Block membrane localization of Ras with farnesylation inhibitors
- Slides: 114