Oncogene Funzione nella cellula normale Cancro Oncogeni Protooncogene












 Cancro")

 SPORADICO (di solito è")







 Genotipo eterozigote per Rb-1 Genotipi delle cellule")

 • Comparative genomic hybridization (CGH) is a fluorescent molecular cytogenetic")











 Normal and tumour DNA samples are")


- Slides: 45
Oncogene Funzione nella cellula normale Cancro
Oncogeni Protooncogene H-ras c-erb. B c-myc c-fos c-kit c-raf RAR- E 6 MDM 2 cicline CDK 2; 4 funzione normale Alterazioni nel cancro Tumori associati
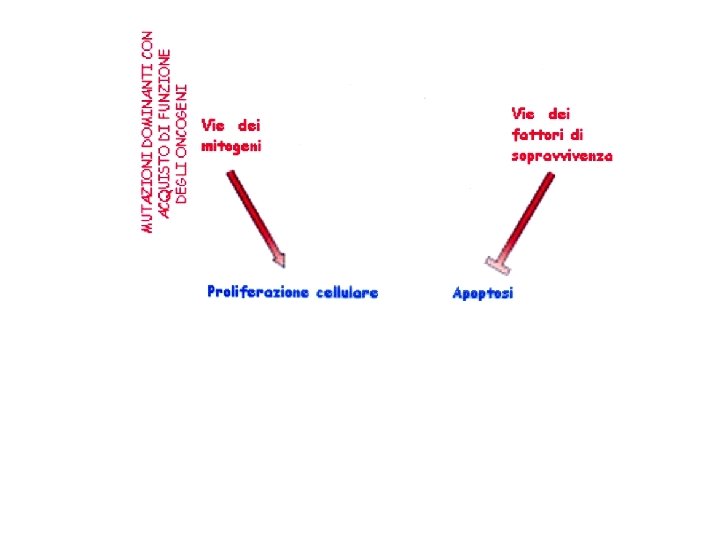
ONCOGENI -> agiscono in maniera dominante; sono causa di forme sporadiche di tumori -Fattori di crescita (GF) -Recettori dei fattori di crescita -Componenti della trasduzione del segnale intracellulare (ras) -Fattori di trascrizione (myc) -Regolatori del ciclo cellulare (cicline)
Meccanismi di attivazione degli oncogeni Differenti tipi di alterazioni genomiche possono avere effetti oncogeni. -Amplificazione genica -Mutazioni puntiformi -Creazione di geni chimerici (traslocazione 9; 22), come BCR-ABL -Traslocazione di geni in regioni cromosomiche trascrizionalmente attive (myc. IGH)
Oltre alle proteine necessarie per l’attivazione della proliferazione cellulare, del ciclo di divisione e dell’apoptosi ce ne sono altre che controllano le proteine attivatrici e ce ne sono ancora altre che sono necessarie a riparare eventuali danni a carico del DNA genomico e contribuire così a prevenire il cancro (ANTIONCOGENI).
ANTIONCOGENI Nel determinare lo sviluppo di un tumore, oltre agli oncogeni possono essere coinvolti geni che specificano proteine che LIMITANO la proliferazione cellulare incontrollata ed i cicli di divisione GENI SOPPRESSORI DEL TUMORE o TSG (sono responsabili sia di forme sporadiche dei tumori che di forme ereditarie) Nei casi di tumore ereditario, in cui sono coinvolti questi geni, un individuo erediterà un allele mutante che inattiva questo tipo di gene che quindi “predispone al tumore” un’alterazione che interviene poi in una cellula somatica nell’allele “sano” rende la cellula omozigote per la mancanza della funzione di quel gene che determinerà quindi una crescita non più regolata della cellula in cui questa alterazione è insorta Si parla in questo caso di “ perdita di eterozigosità” (LOH)
Si può avere la trasformazione cellulare sia per attivazione di un ONCOGENE che per la perdita di funzione di un ANTIONCOGENE Forma sempre attiva anche senza segnali esterni (via della trasduzione del segnale) Ras inattiva GAP NF-1 Ras attiva Ci sono inoltre altre proteine che regolano Ras come NF-1 e GAP (se queste non funzionano ras rimane attiva); queste si comportano come ANTIONCOGENI L’oncoproteina Ras resta bloccata nello stato attivo se c’è la mutazione e il segnale diventa continuo È attivata una serina/treonina chinasi a valle L’interazione con SOS stimola lo scambio GDPGTP
Tumori familiari ed ereditari Circa il 5% di tutti i casi di cancro sono EREDITARI: in questi casi svolge un ruolo decisivo una mutazione germinale Si definisce cancro familiare un tipo di cancro che colpisce numerosi membri di una stessa famiglia e non è necessariamente ereditario Il cancro ereditario invece è un tipo di cancro la cui predisposizione è geneticamente trasmessa e che potrebbe anche essere familiare.
Altre sindromi cancerose EREDITARIE Sindrome cancerosa Cancro primario Posizione cromosomica Designazione del gene Funzione normale del prodotto genico HNPCC Continua……………….
Sindrome cancerosa Cancro primario Posizione cromosomica Designazione del gene Funzione normale del prodotto genico
Albero genealogico di una famiglia con cancro colorettale ereditario non poliposico ( HNPCC) Cancro al colon Altri tipi di tumori Diagnosi non certa C -> cancro al colon S -> cancro allo stomaco E -> cancro all’endometrio P -> cancro al pancreas B -> cancro al tratto urinario Si definiscono famiglie con HNPCC quelle famiglie in cui almeno tre consanguinei su due generazioni hanno avuto una diagnosi di cancro al colon, uno dei quali con diagnosi ad un’età inferiore ai 50 anni
Ipotesi per l’insorgenza dei tumori ereditari È ipotizzabile che attraverso la linea germinale di un individuo si possa ereditare una predisposizione al tumore In questo caso sono interessati geni recessivi che negativamente la crescita cellulare ( gli ANTIONCOGENI) controllano Un esempio di questo tipo di tumore ereditario è fornito dal RETINOBLASTOMA Di questo tipo di tumore se ne conosce una forma ereditaria (che di solito è BILATERALE) ed anche una forma sporadica (che è UNILATERALE) IPOTESI DI KNUDSON: Nella forma familiare si erediterebbe un allele mutato dell’antioncogene (Rb) e si avrebbe una seconda mutazione per varie cause ( una forma di perdita di eterozigosità). La predisposizione al tumore è DOMINANTE e lo sviluppo del tumore è recessivo
Alberi genealogici per il retinoblastoma EREDITARIO (di solito è bilaterale) SPORADICO (di solito è unilaterale)
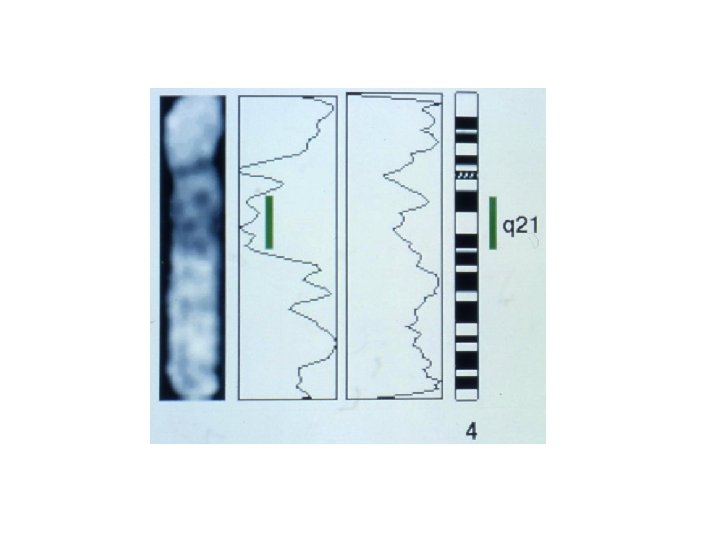
Dimostrazione dell’ ipotesi di Knudson Rari bambini con retinoblastoma bilaterale mostravano anche altri gravi difetti e ritardo mentale Citologicamente questi bambini mostravano una delezione nel braccio lungo del cromosoma 13 nella regione 13 q 14 (regione minima deleta >SRO)
Mappatura del gene del retinoblastoma p SRO q
Dimostrazione dell’ ipotesi di Knudson Rari bambini con retinoblastoma bilaterale mostravano anche altri gravi difetti e ritardo mentale Citologicamente questi bambini mostravano una delezione nel braccio lungo del cromosoma 13 nella regione 13 q 14 (regione minima deleta >SRO) In questa regione mappa l’esterasi D che è un enzima costitutivo; si è quindi effettuata un’analisi di linkage con l’esterasi D È stata effettuata l’analisi dell’attività enzimatica dell’esterasi D Nei casi di RETINOBLASTOMA EREDITARIO l’attività dell’esterasi D risulta di 0, 5 rispetto a quella di un individuo normale nel sangue dell’individuo che mostra il tumore nella retina ed è ridotta a 0 nelle cellule tumorali (della retina) -> si è avuta la PERDITA’ DI ETEROZIGOSITA’. Nei casi di RETINOBLASTOMA SPORADICO nel sangue degli individui affetti l’attività enzimatica dell’esterasi D è pari ad 1, mentre nelle cellule tumorali della retina è pari a 0
Ipotesi di Knudson Retinoblastoma EREDITARIO X X X Retinoblastoma NON EREDITARIO Occasionalmente in una cellula della retina avviene una mutazione a livello del gene Rb Gene mutato per Rb XX X X Occasionalmente in una cellula della retina avviene una SECONDA mutazione a livello del gene Rb Si ha quindi una proliferazione incontrollata di questa cellula che risulta nella formazione del retinoblastoma (BIILATERALE) La maggior parte degli individui che ereditano Rb-1 in eterozigosi sviluppano il tumore X X La seconda copia del gene è molto raramente inattivatta nella STESSA cellula Si ha quindi una proliferazione incontrollata di questa cellula che risulta nella formazione del retinoblastoma (UNILATERALE) Solo in 1/30000 persone si sviluppa questo tipo di tumore spontaneamente
Retinoblastoma familiare Retinoblastoma sporadico
Strategia per il clonaggio del gene del retinoblastoma 5 kb Chromosome jumping Esterasi-D Rb-1 (1, 5 kb) Northern su RNA da retina 5 kb c. DNA di Rb-1 1 kb R 180 kb -> 17 esoni m. RNA 3000 nt R B B R
Proteina p. RB 1 Serina 252* Treonina 249* 928 608 356 373* 788 795 821 811* 612 807* La proteina è di 928 amminoacidi, è ricca di prolina e glicina e controlla il ciclo cellulare (passaggio dalla fase G 1 alla fase S) Nel nucleo, durante la fase G 1, p. RB interagisce proprio con un fattore di trascrizione E 2 F e lo INATTIVA. Quando la cellula passa da G 1 alla fase S, si forma un complesso CDK 4/ciclina D 1, questo complesso FOSFORILA p. RB e quindi a seguito della fosforilazione E 2 F viene rilasciato e diventa ATTIVO trascrizionalmente. La fosforilazione di p. RB è transitoria; appena i complessi CDK/ciclina sono degradati e la cellula si muove attraverso il ciclo cellulare alla fase G 1 iniziale, la fosforilazione di p. RB decade.
Meccanismi per la perdita di eterozigosità (LOH) Genotipo eterozigote per Rb-1 Genotipi delle cellule della retina di questo individuo prive della funzione Rb Genotipo omozigote per Rb-1 per SECONDA MUTAZIONE Genotipo omozigote per Rb-1 per PERDITA CROMOSOMICA oppure PERDITA CROMOSOMICA + NON DISGIUNZIONE Genotipo per DELEZIONE che manifesta fenotipo Rb-1 Genotipo omozigote per Rb-1 per RICOMBINAZIONE MITOTICA
Conclusioni 1 I geni coinvolti nell’insorgenza dei tumori sono raggruppati in due grandi categorie: ONCOGENI -> geni che controllano la proliferazione cellulare, la progressione attraverso il ciclo cellulare e l’apoptosi La maggior parte di questi geni se mutati (anche in una sola copia) possono determinare l’insorgenza del tumore (Mutazioni Dominanti) ANTIONCOGENI -> geni che regolano i geni coinvolti nella proliferazione cellulare, la progressione attraverso il ciclo cellulare e l’apoptosi Per l’induzione dei tumori da parte di questi geni è necessario che entrambe le copie del gene (sui due cromosomi omologhi) siano mutate (Mutazioni recessive, necessitano della perdita dell’eterozigosità. Per i tumori ereditari questo si può realizzare in un unico passaggio in quanto si eradita già una copia mutata del gene, nei tumori sporadici la cellula prima deve diventare eterozigote per la mutazione e poi deve acquistare la perdita dell’eterozigosità
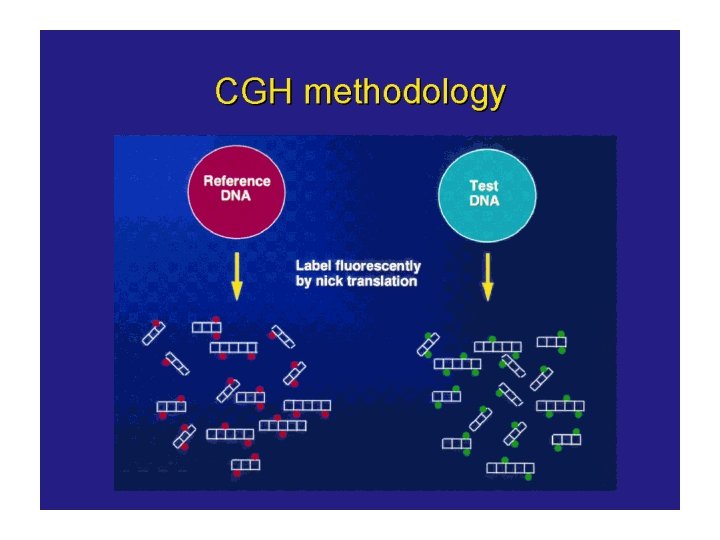
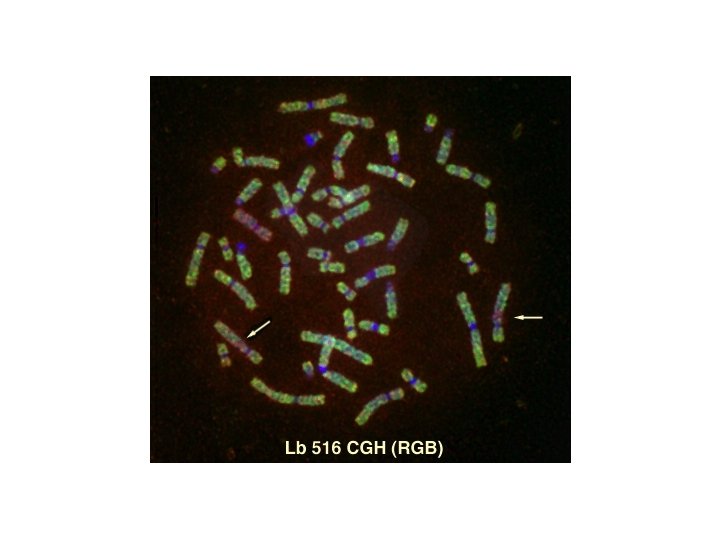
Comparative Genomic Hybridization (CGH) • Comparative genomic hybridization (CGH) is a fluorescent molecular cytogenetic technique that identifies DNA gains and losses, mapping these variations to normal metaphase chromosomes. • It is a powerful tool for screening chromosomal copy number changes in tumor genomes and has the advantage of analyzing entire genomes within a single experiment. • It is particularly applicable to the study of tumors which do not yield sufficient metaphases for cytogenetic analysis and can be applied to fresh or frozen tissues, cell lines, and archival formalin-fixed paraffin-embedded samples.
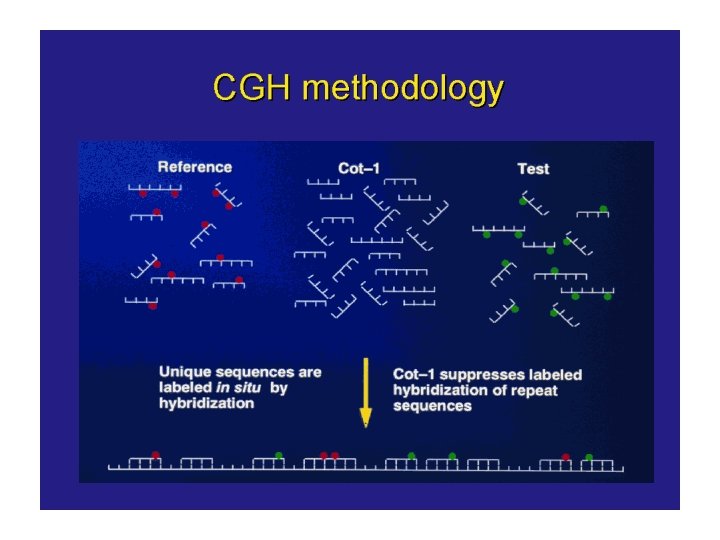
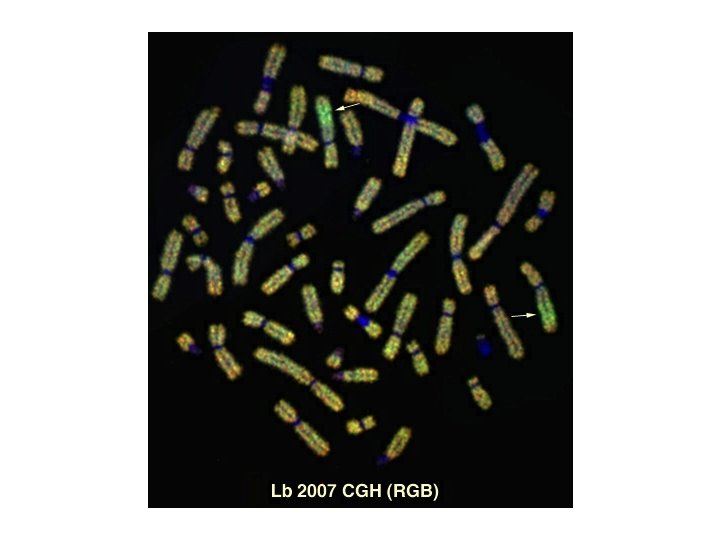
CGH is based on quantitative two-color fluorescence in situ hybridization. Equal amounts of differentially labeled tumor genomic DNA and normal reference DNA are mixed together and hybridized under conditions of Cot-1 DNA suppression to normal metaphase spreads. The labeled probes are detected with two different fluorochromes, e. g. , FITC o CY 3 for tumor DNA and TRITC o CY 5 for the normal DNA. The difference in fluorescence Intensities along the chromosomes in the reference metaphase spread are a reflection of the copy number changes of corresponding sequences in the tumor DNA.
DNA da testare
DAPI
FITC
TRITC
DAPI, FITC, TRITC superposition
CGH has the advantage of requiring only genomic tumor DNA, making it highly useful for cancer cytogenetics, circumventing the need for high quality tumor metaphase spreads. The ability to study archival material allows retrospective analysis which can correlate chromosomal aberrations with the clinical course. Since its introduction in 1992, CGH has been applied to a broad variety of tumor types which have previously defied comprehensive cytogenetic analysis by traditional methods. CGH has, for example: • Revealed consistent genetic imbalances and multiple amplification sites in carcinomas of the brain, colon, prostate, cervix, and breast. For instance: it identified chromosome 7 gain and chromosome 10 loss as landmark aberrations in glioblastomas, and specific gains of chromosomes 1, 8, 17, and 20 and loss of 13 q and 17 p in breast cancer. • Found chromosomal aberrations in human leukemia, lymphoma, and solid tumors has identified non-random tumor and tumor-stage specific genetic changes. This information can guide positional cloning efforts. • Become an important initial screening test for chromosomal gains and losses in solid tumor progression, and the results derived from these experiments can be applied to the development of more specific diagnostics.
array-CGH Extract, amplify and label DNA CGH Hybridize to metaphase spread on BAC array
Array-CGH greatly improves the resolution of the CGH technique by substituting the hybridization target, the metaphase chromosome spread, with genomic segments spotted in an array format. Resolution of the techinque: • traditional CGH : 10 Mb • array-CGH: 75 -135 kb
DNA da testare
General principles of array comparative genomic hybridization. (a) Normal and tumour DNA samples are isolated and used to create fluorescently labelled probes (FITC and TRITC). The probes are pooled and competitively cohybridized to a glass slide spotted with a known array of mapped genomic clones (BAC or PAC clones). The arrays are analysed with a microarray scanner, producing an image that is used to assess the ratios of the FITC to TRITC intensities for each clone. (b) A ratio profile is assembled to determine relative copy number changes between the reference and tumour samples. Each dot on the graph represents a clone. Values to the left of the ‘ 0’ line indicate a loss of a genomic region, values to the right indicate a gain or amplification, and values at ‘ 0’ indicate no change.
Whole-genome profile of a cell lung cancer sample. Each dot represents a BAC clone and is visualized using See. GH software. Clones around the centre line indicate normal regions in the tumour genome in relation to the normal reference sample. Clones to the left of centre represent homozygous or hemizygous deletions in the tumour, whereas clones to the right represent gains and amplifications. Focal amplifications of common oncogenes are magnified.