OCUKLUK AI SIK GRLEN GENETK HASTALIKLAR HASTALIKLARIN ETYOLOJSNDE




 - Trizomi 18 -")








. • Evlilik öncesi")
 Tip 2 - Ara tip")
 Genetik Danışma: Otozomal")

 Genetik Tanı: Mutasyon analizi Genetik Danışma: Otozomal resesif. Hasta")
 Su ve tuz kaybı ile giden (ağır form)")



 Genetik")



- Slides: 27
ÇOCUKLUK ÇAĞI SIK GÖRÜLEN GENETİK HASTALIKLAR
HASTALIKLARIN ETYOLOJİSİNDE GENETİK NEDENLER 1. TEK GEN hastalıkları 2. KROMOZOM anomalileri 3. MULTİFAKTÖRYEL hastalıklar
MULTİFAKTÖRYEL HASTALIKLAR KONJENİTAL ANOMALİLER’in çoğu multifaktöryeldir. Çocukluk çağı genetik hastalıkların en büyük grubu multifaktöryel hastalıklardır.
KROMOZOMAL HASTALIKLAR Kromozom veya genom mutasyonları sonucu oluşan hastalıklardır. Hemen hepsinde zeka geriliği ve dismorfik bulgular gözlenir.
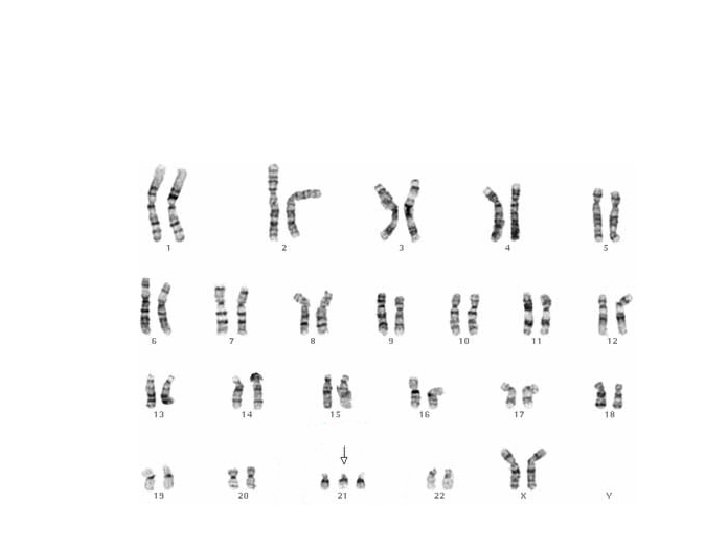
Sık Görülen Kromozomal Hastalıklar Otozomal - Down Sendromu (Trizomi 21) - Trizomi 18 - Trizomi 13 Gonozomal - 45, X Turner Sendromu -47, XXY Klinefelter Sendromu - 47, XYY - 47, XXX
DOWN Sendromu Trizomi 21 En sık görülen kromozom anomalisi Zeka geriliklerinin en sık görülen genetik nedeni İleri anne yaşı Tanı %95 klinik bulgularla Mozaik formlarında zeka geriliği daha hafif değildir!!!
Trizomi 18 -Edward Sendromu 3/1000 Mikrosefali, yarık damak dudak Konjenital kalp hastalıkları Clench hand, rocker bottom feet. Ağır gelişme ve zeka geriliği.
Trizomi 13 -Patau Sendromu 1/5000 ◦ ◦ ◦ Scalp defektleri, Yarık damak dudak (60 -80%), Holoprosensefali Konjenital kalp hastalığı 80% (VSD, PDA, and ASD) Omfalosel Polidaktili Ağır zeka geriliği
TEK GEN mutasyonlarına bağlı hastalıklar DNA daki değişiklik sonucu tek bir genin işlevinin bozulması ile ortaya çıkarlar. Genelde Mendel kalıtım örneğine uygun kalıtılırlar. OD, OR , X’e bağlı Bazıları Non Mendelian kalıtım gösterirler. UPD, İmprinting Germinal mozaisizm Dinamik mutasyon Mitokondrial kalıtım
Tek Gen Hastalıkları Sık Görülen Tek Gen Hastalıkları Nadir Tek Gen Hastalıkları Sayıları az (onlarla) Görülme oranları yüksek (1/500 -1/10 000) Sayıları çok fazla ( binlerle) Görülme oranları çok düşük (1/20 000 - 1/100. 000)
Sık Görülen Tek Gen Hastalıkları Otozomal Dominant - Akondroplazi - Marfan S. - Nörofibromatozis - Osteogenezis imperfekta Otozomal Resesif - Beta. Talasemi - Kistik Fibroz - Fenilketonuri - Spinal kas Atrofisi - Konjenital Adrenal hiperplazi -Mukopolisakkaridozlar
AKONDROPLAZİ Tanı : Orantısız boy kısalığı + radyolojik bulgular Genetik Tanı : G 380 R mutasyonu Prenatal Tanı : G 380 R mutasyonu Genetik Danışma : OD Hasta çocuğun ebeveyni normal ise yineleme riski düşük. Hastanın çocukları için Akondroplazi olma riski % 50
BETA TALASEMİ Tanı : Klinik + Hb elektroforezi Otozomal resesif. Her iki ebeveyn taşıyıcı ise veya hasta çocukları varsa her gebelikte yineleme riski %25 dir. Hasta çocuğun sağlıklı kardeşleri ile ebeveynin kardeşleri, yeğen ve kuzenlerinin taşıyıcılık riskleri artmıştır. Taşıyıcı Testi: Hemoglobin A 2 düzeyi Genetik Tanı : Mutasyon analizi Prenatal tanı testi: Bilinen mutasyon aranır Mutasyon bilinmiyorsa Bağlantı analizi yapılır
BETA TALASEMİ Türkiyede Sorunlar • Türkiyede taşıyıcı sıklığı çok yüksek (1/23). • Evlilik öncesi Talasemi tarama testi yaygın değil. • Hasta çocuğun ailesi ve yakınlarına yineleme riski ve prenatal tanı hakkında yeterli bilgi verilmiyor. • Riskli ailelerde moleküler testlerin gebelik öncesi tamamlanması gerekiyor.
Spinal Kas Atrofisi TİP 1 (Werdnig –Hoffmann H. ) Tip 2 - Ara tip Genel hipotoni, kas güçsüzlüğü, DTR negatif, mentali normal
Spinal Kas Atrofisi Tanı: Klinik bulgular + Kas bx, EMG (invazif) Genetik Danışma: Otozomal resesif. Hasta çocuk varsa her gebelikte yineleme riski % 25 Genetik Tanı: Klinik + SMNt geninde delesyon (Hastaların % 98’inde saptanır) (noninvazif test) Prenatal Tanı : SMNt geninde delesyon aranması
KİSTİK FİBROZ Tanı: Klinik bulgular + pozitif ter testi Genetik Tanı: Mutasyon analizi Genetik Danışma: Otozomal resesif. Her gebelikte yineleme riski % 25 Prenatal tanı: Bilinen mutasyon aranır Mutasyon bilinmiyorsa bağlantı analizi Hasta çocuğun mutasyonu bilinmeden yeni bir gebelik oluşması prenatal tanıda sorun yaratır. Çok sayıda mutasyon olduğundan inceleme uzun sürebilir.
FENİLKETONURİ Tanı: Tarama testi (Hiperfenilalaninemi) Genetik Tanı: Mutasyon analizi Genetik Danışma: Otozomal resesif. Hasta bir çocuğu olan anne ve baba zorunlu taşıyıcıdır ve izleyen her gebelikte risk % 25. Prenatal Tanı: İndeks hastanın mutasyonu aranır. Bağlantı analizi yapılır.
KONJENİTAL ADRENAL HİPERPLAZİ (21 hidroksilaz eksikliği) Su ve tuz kaybı ile giden (ağır form) Basit virilizan tip (kuşkulu genitalya)
KONJENİTAL ADRENAL HİPERPLAZİ Tanı : Klinik + serum 17 -hidroksiprogesteron /androstenedion düzeyleri Genetik Tanı : Mutasyon analizi Genetik Danışma : Otozomal resesif. Hasta bir çocuktan sonra yineleme riski % 25. Prenatal Tanı: İndekste mutasyon analizi Prenatal Tedavi: Riskli gebelere 5. GH da fetusun tanısı kesinleşinceye kadar sürdürülen deksametason tedavisi başlanır , fetus kız ve hasta ise tedavi gebelik boyunca sürdürülür.
Sık Görülen Tek Gen Hastalıkları X’e Bağlı Resesif Duchenne Kas D/BMD Hemofili A / B X’e Bağlı Dominant Rett sendromu D vitaminine bağlı raşitizm Non Mendel kalıtım gösterenler Frajil-X Sendromu-Dinamik mutasyon Leber optik Atrofi –Mitokondrial kalıtım Prader Willi Sendromu- UPD/İmprinting
Duchenne Kas Distrofisi Motor gelişme geriliği, anormal yürüme, sık düşme, Gowers bulgusu, baldır kaslarında belirginlik, Serum CPK artış, pozitif aile öyküsü
Duchenne Kas Distrofisi Tanı : Klinik + Kas biyopsisinde distrofin yokluğu (İnvazif test) Genetik Tanı: Distrofin gen delesyonu (% 55 -65) Rutin test Nokta mutasyon, dup, insertion (%35) Araştırma Genetik Danışma : Hasta çocuğun anne ve anne tarafındaki kadın akrabalarının taşıyıcılık riski artmıştır Taşıyıcı tanısı: FISH yöntemi / moleküler analiz Prenatal Tanı: Distrofin gen delesyonu bakılır Delesyon yoksa bağlantı analizi yapılabilir.
HEMOFİLİ A Tanı : Klinik + Faktör 8 eksikliği Genetik Danışma : X’e bağlı resesif. Hasta kişinin annesi ve anne tarafındaki kadınlarda taşıyıcılık riski artmıştır. Taşıyıcı tanısı: Faktör 8 düzeyi Genetik Tanı : Mutasyon analizi Prenatal Tanı: İndekste mutasyonun belirlenmesi Bağlantı analizi
Frajil X • Erkek çocuklarda mental geriliğin Down Sendromundan sonraki 2. nedeni • Mendel kalıtımına uymayan bir tek gen hastalığı • 3’lü nükleotit tekrar artışına bağlı gelişiyor.
Tanı: Klinik bulgular Genetik Tanı: Klinik +CGG tekrar sayısında artış Normal 45 -55 tekrar Premutasyon 55 -200 tekrar (taşıyıcı) Full mutasyon >200 tekrar Prenatal Tanı : CGG tekrar sayısına bakılması