Normal mode analysis NMA tutorial and lecture notes
 tutorial and lecture notes by K. Hinsen Serkan Apaydın")


 0 Rmin Conformation (r)")
 =0. 5 (r − Rmin)’ · K(Rmin)")
 =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)")
 =0. 5 (r − Rmin)’ · K(Rmin) ·")
 =0. 5 (r − Rmin)’ ·")
 U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)")









 • Capture collective motions – Specific to a protein")
 f can be a step function or may have an")
 All atom or C-alpha based models… • Or a step")
 Gaussian network model:")



 <r> = R <(r − R)'> =k. BT inv(K) Angel E. García,")
 • Cannot capture the fine level intricacies of the motion • Freezing")

 • Explaining which modes/frequencies are involved in a particular")



")
 http: //www. bioinfo. no/tools/normalmodes")
")
: http: //ignm. ccbb. pitt. edu/")
")
")




















")

 and closed (R) structures of HIV protease")



 • Protein decomposed into rigid clusters • Better than ad-hoc")


 Way of generating new conformations? Flexibility/mobili ty index")



- Slides: 73
Normal mode analysis (NMA) tutorial and lecture notes by K. Hinsen Serkan Apaydın
Protein flexibility Frequency spectrum of a protein Over half of the 3800 known protein movements can be modelled by displacing the studied structure using at most two low-frequency normal modes. Gerstein et al. 2002
Outline • NMA – What it is – Vibrational dynamics – Brownian modes – Coarse grained models – Essential dynamics
Harmonic approximation Energy (U) 0 Rmin Conformation (r)
Harmonic approximation U 0 Rmin r U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)
NMA U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)
NMA Normal mode direction 1 U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)
NMA Normal mode direction 2 -e 2 U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin)
NMA (2) U(r) =0. 5 (r − Rmin)’ · K(Rmin) · (r − Rmin) min O(n 3)
Properties of NMA • The eigenvalues describe the energetic cost of displacing the system by one length unit along the eigenvectors. • For a given amount of energy, the molecule can move more along the low frequency normal modes • The first six eigenvalues are 0, corresponding to rigid body movements of the protein
4 ways of doing NMA A. Using minimization to obtain starting conformation, and computing the Hessian K: 1. 2. Vibrational NMA Brownian NMA B. Given starting structure: 1. Coarse grained models C. Given set of conformations corresponding to the motion of the molecule: 1. Essential Dynamics
1. Vibrational NMA • derived from standard all -atom potentials by energy minimization • time scale: < residence time in a minimum • appropriate for studying fast motions • Useful when comparing to spectroscopic measurements • Requires minimization and Hessian computation
1. Vibrational NMA
Vibrational frequency spectrum
2. Brownian NMA • derived from standard all -atom potentials by energy minimization • time scale: > residence time in a minimum • appropriate for studying slow motions • Requires minimization and Hessian computation
2. Brownian NMA
The friction coefficients • describe energy barriers between conformational substates • Can be obtained from MD trajectories (<xi 2>) • Depend on local atomic density (not a solvent effect) http: //dirac. cnrs-orleans. fr/plone/Members/hinsen/
3. Coarse grained models • Around a given structure • time scale: >> residence time in a minimum • appropriate for studying slow, diffusive motions (jump between local minima) • Does not require expensive minimization and Hessian computation
3. Coarse grained models (2) • Capture collective motions – Specific to a protein – Usually related to its function – Largest amplitudes • Atoms are point masses • Springs between nearby points
Coarse grained models (3) f can be a step function or may have an exponential dependence. Elastic network model NMA (aka ANM) Find Hessian of V, then eigendecomposition Gaussian network model
Coarse grained models (4) All atom or C-alpha based models… • Or a step function…
Equilibrium fluctuations Ribonuclease T 1 Disulphide bond facilitator A (Dsb. A) Gaussian network model: Theory and applications. Rader et al. (2006)
Difference between ENM NMA and GNM • GNM more accurate in prediction of meansquare displacements • GNM does not provide the normal mode directions
Lower resolution models • Groups of residues clustered into : • unified sites G Li, Q Cui - Biophysical Journal, 2002 • Rigid blocks (rotation and translation of blocks (RTB) model) • To examine larger biomolecular assemblies
4. Essential dynamics • Given a set of structures that reflect the flexibility of the molecule • Find the coordinates that contribute significantly to the fluctuations • time scale: >> residence time in a minimum
Essential Dynamics(2) <r> = R <(r − R)'> =k. BT inv(K) Angel E. García, Kevin Y. Sanbonmatsu Proteins. 2001 Feb 15; 42(3): 345 -54.
Essential dynamics(3) • Cannot capture the fine level intricacies of the motion • Freezing the small dofs make small energy barriers insurmountable • Need to run MD for a long time in order to 38, 150, 199 dofs obtain sufficient samples
Applications of normal modes • Use all modes or a large subset – Analytical representation of a potential well – Limitations: • approximate nature of the harmonic approximation • Choice of a subset • Properties of individual modes – Must avoid overinterpretation of the data • E. g. , discussing differences of modes equal in energy • No more meaningful than discussing differences between motion in an arbitrarily chosen Cartesian coord. system
Applications of normal modes (2) • Explaining which modes/frequencies are involved in a particular domain’s motion • Answered using projection methods: – Normal modes form a basis of the config. space of the protein – Given displacement d, pi = d · ei • Contribution of mode i to the motion under consideration – Cumulative contribution of modes to displacement
Cumulative projections of transmembrane helices in Ca. ATPase
Comparison chart Amplitude Time Starting scale structure Vibration Small Short By al Minimization Brownian Large Long By Minimization Coarse Large Long Given grained Essential Large Long Given Practical N Y/N Y N
Summary NMA: • no sampling problem • computational efficiency, especially for coarse-grained models • simplicity in application • Predicts experimental quantities related to flexibility, such as B-factors, well.
• http: //igs-server. cnrs-mrs. fr/elnemo/ (all atom)
Web. NM: (C-alpha based) http: //www. bioinfo. no/tools/normalmodes
http: //promode. socs. waseda. ac. jp/pages/jsp/index. jsp (all-atom)
Ignm (C-alpha based): http: //ignm. ccbb. pitt. edu/
http: //molmovdb. org/nma/ (C-alpha based)
http: //lorentz. immstr. pasteur. fr/nomad-ref. php (all atomic or just C-alpha)
Protein Flexibility Predictions Using Graph Theory Jacobs, Rader, Kuhn and Thorpe Proteins: Structure, function and genetics 44: 150 -165 (2001) Serkan Apaydın
Characterizing intrinsic flexibility and rigidity within a protein 1. Compares different conformational states Limited by the diversity of the conformational states
Characterizing intrinsic flexibility and rigidity within a protein 2. Simulates molecular motion using MD Limited by the computational time
Characterizing intrinsic flexibility and rigidity within a protein 3. Identifies rigid protein domains or flexible hinge joints based on a single conformation Can provide a starting point for more efficient MD or MCS
Outline • • The main idea: constraint counting Brute force algorithm Rigidity theory Pebble game analysis Rigid cluster decomposition Flexibility Index Examples
Overview of FIRST • Floppy Inclusion and Rigid Substructure Topography • Given constraints: – Covalent bonds – hydrogen bonds – Salt bridges • Evaluate mechanical properties of the protein: Find regions that are: – rigid – move collectively – move independently of other regions Compute a relative degree of flexibility for each region
Rigidity in Networks – a history • • introduces constraints on the motions of mechanical systems 1864: Maxwell determined whether structures are stable or deformable 1788: Lagrange applications in engineering, such as the stability of truss configurations in bridges • 1970: Laman’s theorem: determines the degrees of freedom within 2 D networks and allow rigid and flexible regions to be found extended to bond-bending networks in 3 D http: //unabridged. m-w. com
Brute force algorithm to test rigidity INDEPENDENT ORACLE REDUNDANT
Brute force algorithm to test rigidity INDEPENDENT ORACLE REDUNDANT • Compute normal modes w/ and w/o the constraint Complexity? O(n 2. n 3) O(n 5) • If the number of zero eigenvalues remains constant, then the constraint is redundant.
Laman’s theorem accelerates constraint counting • Constraint counting to all the subgraphs – Applying directly, complexity is O(exp(n)) – Applying recursively, pebble game algorithm. Complexity is O(n 2), O(n) in practice.
Pebble Game • 3 pebbles per node • Each edge must be covered by a pebble if it is independent • Pebbles remaining with nodes are free and represent DOFs of the system • An edge once covered should stay covered but pebbles can be rearranged.
The Pebble Game: A Demonstration Mykyta Chubynsky and M. F. Thorpe Arizona State University
Pebble game Flexible hinges Hyperstatic
Pebble game
Final arrangement of pebbles In 2 D, 2 pebbles / node. Blue: Free pebble, one DOF Red: Associated with an edge, a “used” DOF by the constraint This arrangement determines the flexible regions and rigid clusters
Finding rigid clusters • A rigid cluster can have a maximum of 3 pebbles in 2 D • Rearrange the pebbles to obtain > 3 pebbles in a connected region
Finding rigid clusters This is not a rigid cluster since there are 4 pebbles here
Hydrogen bonds • Selection of a cut-off energy for hydrogen bonds • Selected based on agreement of hydrogen bonds within a family of protein structures
Hydrogen bond energy computation r <=2. 6 Å 90 <= 180 d<= 3. 6 Å V 0 = 8 kcal/mol d 0 = 2. 8 Å sp 3 donor-sp 3 acceptor F=cos 2 cos 2( -109. 5)
Flexibility Index • 4 -3 =1 DOF • 3 rotatable bonds • F = 1/3 • 1 redundant constraint • 6 distance constraints • F = -1/6 – #(independent DOFs)/#(rotatable bonds) – #(redundant constraints)/#(distance constraints)
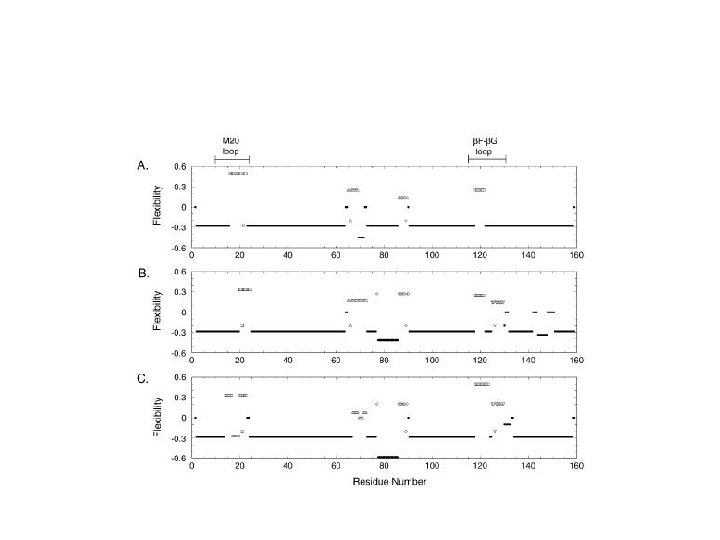
Application to HIV protease (unbound)
Agreement with experiment
Comparison of the open (L) and closed (R) structures of HIV protease
Dihydrofolate reductase
Rigid cluster decomposition barnase Maltodextrin binding protein Gohlke and Thorpe. Biophysical Journal 91: 2115 -2120 (2006)
FIRST/FRODA predictions barnase Maltodextrin binding protein Gohlke and Thorpe. Biophysical Journal 91: 2115 -2120 (2006)
Rigid cluster NMA (RCNMA) • Protein decomposed into rigid clusters • Better than ad-hoc definition of blocks • Rotation-Translation Block Analysis for the resulting network • 9 -27 times less memory • 25 -125 times faster
Comparison of RCNMA w/ ENM Barnase r 2 0. 56 vs. 0. 50 Maltodextrin binding protein r 2 0. 62 vs. 0. 55 Gohlke and Thorpe. Biophysical Journal 91: 2115 -2120 (2006)
Comparison of FIRST and NMA Allatom? Speed? Given starting pt. Freq. spectrum? FIRST Y Y Y Low frequency motion NMA Y / N (N Y (N for coarse VNMA, grained) Brownian, Y All frequencies ED)
Comparison of FIRST and NMA (2) Way of generating new conformations? Flexibility/mobili ty index FIRST Y (with ROCK or Y* FRODA) NMA Y Y *: incorrect for rigid regions flanked by flexible hinges
http: //flexweb. asu. edu
Conclusion • • • Rigidity theory Constraint counting Based on a single structure Fast Available on the web: http: //flexweb. asu. edu • Tools using FIRST to generate new conformations: ROCK, FRODA