Next Generation Sequencing Sample fragmentation Library preparation Sequencing

高通量测序技术简介 Next Generation Sequencing • • Sample fragmentation Library preparation Sequencing reaction Data analysis Roche 454 焦磷酸测序 Pyrophosphate Sequencing Illumina Solexa 合成测序 Sequence by Synthesize ABI SOLi. D 连接法测序 Sequence by Ligation

Roche 454 焦磷酸测序 Pyrophosphate Sequencing 基本原理
 A + PCR Reagents + Emulsion Oil B")
454 sequencing: Emulsion PCR (em. PCR) A + PCR Reagents + Emulsion Oil B Micro-reactors Adapter carrying library DNA Mix DNA Library & capture beads (limited dilution) Create “Water-in-oil” emulsion Adapter complement Enrich Anneal Seq primer “Break micro-reactors” Isolate DNA containing beads Perform emulsion PCR § Generation of millions of clonally amplified templates on each bead § No cloning and colony picking

454 sequencing: Deposition of DNA beads into the Pico. Titer™Plate Load beads into Pico. Titer™Plate Load Enzyme Beads Centrifuge Step

Illumina Solexa 合成测序 Sequence by Synthesize 基本原理

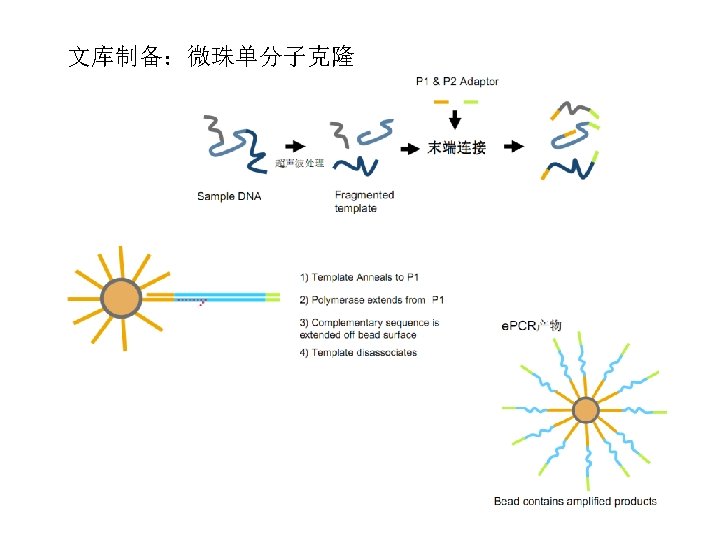
Clonal Single Molecule Arrays 单分子克隆 Attach single molecules to surface Amplify to form clusters Prepare DNA fragments Ligate adapters Sequence ~1000 molecules per ~ 1 µm cluster ~1000 clusters per 100 µm square ~40 million clusters per experiment 20 microns

Reversible Terminator Chemistry 可逆终止反应 • All 4 labelled nucleotides in 1 reaction O cleavage fluor site HN O O N DNA O PPP 3’ HN 5’ O block Incorporation Detection Deblock; fluor removal O N O 3’ OH free 3’ end Next cycle X
 3’ 5’ Cycle 1: Add sequencing reagents First base incorporated Remove unincorporated")
Sequencing-by-Synthesis (SBS) 3’ 5’ Cycle 1: Add sequencing reagents First base incorporated Remove unincorporated bases A T G C C G T Cycle 2 -n: Add sequencing reagents and repeat T A C A Detect signal C G A T T A G A C T C C G A G C T C G A T 5’ 1、每轮测序反应加入四种带有荧光标记的d. NTP,末端带有可 以被去除的阻断基团 2、每轮反应只能整合一个核苷酸,仪器读取相应的荧光信号 3、信号读取结束,用化学方法去除阻断基团,进行下一轮测序 反应

Base calling from the raw data TGCTACGAT… 1 2 3 4 5 6 7 8 TTTTTTTGT… The identity of each base of a cluster is read off from sequential images 根据每个点每轮反应读取的荧光信号序列,转换成相 应的DNA序列 9

Solexa 测序 Workflow

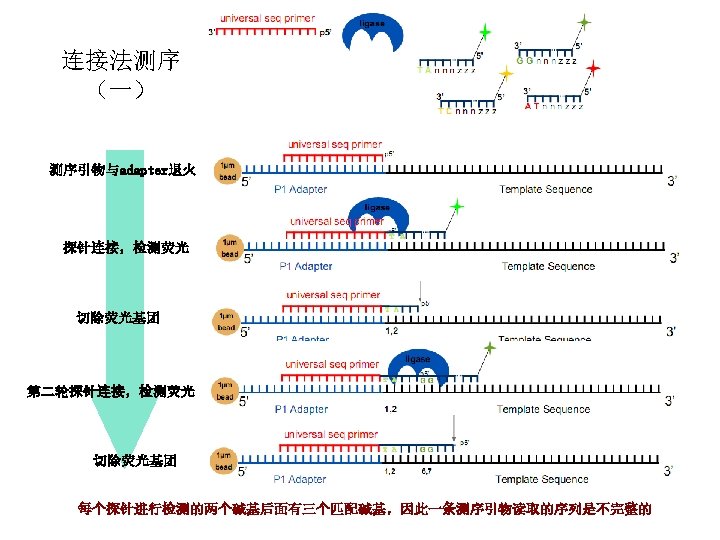
ABI SOLi. D 连接法测序 Sequence by Ligation 基本原理




 转录组深度测序(transcriptome re-sequencing) Digital expression profiling")
高通量测序的应用 • • • De novo 测序 基因深度测序(genome re-sequencing) 转录组深度测序(transcriptome re-sequencing) Digital expression profiling Ch. IP-seq Methy-seq
 :恶性胸膜间皮瘤 pulmonary adenocarcinoma (ADCA):肺腺癌")
Transcriptome resequencing: malignant pleural mesotheliomas (MPMs) :恶性胸膜间皮瘤 pulmonary adenocarcinoma (ADCA):肺腺癌

Transcriptome characteristics Expression difference between MPM and ADCA sample compare to a lung tissue control Solid line: at least one read Dashed line:at least 20 reads Analysis of percentage of reads containing known coding region SNVs in the six tissue samples. SNV: Single Nucleotide Substitution Variant
: 人大脑组织与UHR(Universal Human Reference)的表达差异")
Digital expression profiling(1): 人大脑组织与UHR(Universal Human Reference)的表达差异

Digital expression profiling & micro. RNA re-sequencing: h. ESC: human embryonic stem cells EB: embryoid bodies
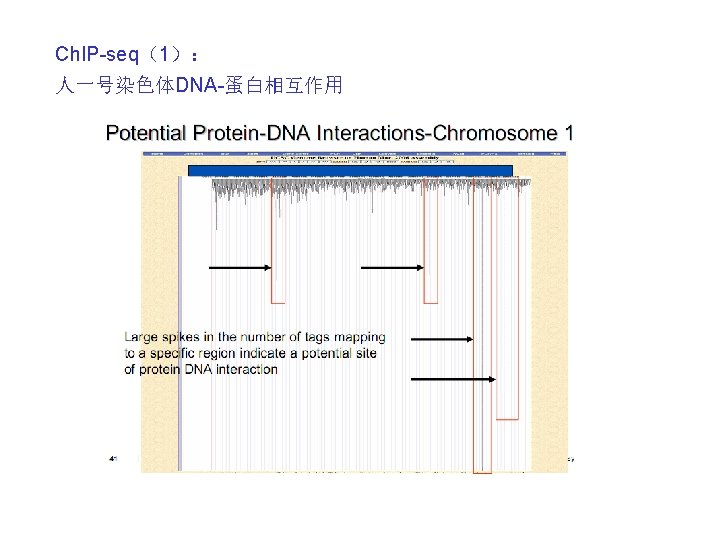
: Sequenced short reads (typically � 25– 50 bp) from Ch. IP-Seq experiments")
Ch. IP-seq(2): Sequenced short reads (typically � 25– 50 bp) from Ch. IP-Seq experiments are first mapped onto the reference genome. The mapped reads are then used to estimate statistical parameters, which include the estimation of the average length F of sequenced DNA fragments.
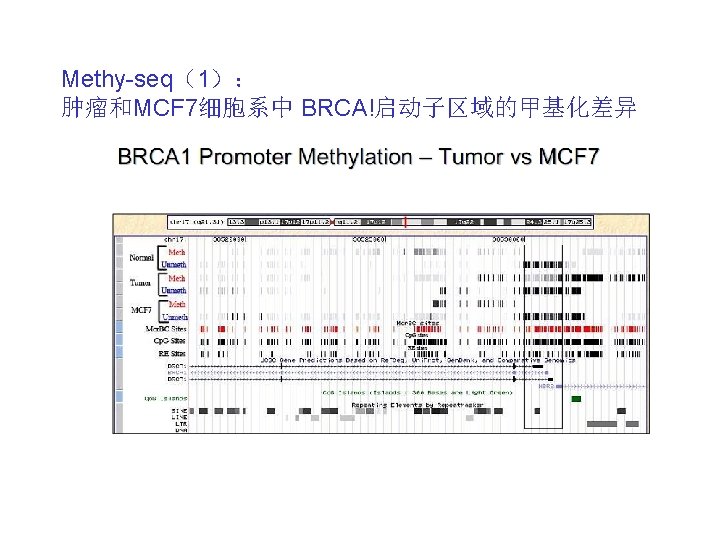
: Some highlights: Correlation between Ch. IP-Seq and his prior SAGE-like method (called GMAT)")
Methy-seq(2): Some highlights: Correlation between Ch. IP-Seq and his prior SAGE-like method (called GMAT) has r=0. 906 ‘However the resolution with Ch. IP-Seq was dramatically higher. Furthermore, Ch. IP-Seq was more sensitive and generated less falsenegative regions’ 12, 726 genes whose transcription levels are known in CD 4+ T-cells were correlated with the histone modifications and 35, 961 Pol II binding site ‘islands’ were identified ‘This cost-effective method produces digital-quality data and should find broad applications in our efforts to understand the contribution of the human epigenomes in gene expression and epigenetic inheritance’

部分参考文献阅读 Genome re-sequencing • • van Orsouw N J, Hogers R C, Janssen A, et al. Complexity reduction of polymorphic sequences (CRo. PS): a novel approach for large-scale polymorphism discovery in complex genomes. PLo. S ONE, 2007, 2(11): e 1172 Hillier L W, Marth G T, Quinlan A R, et al. Whole-genome sequencing and variant discovery in C. elegans. Nat Methods, 2008, 5(2): 183— 188 Transcriptome re-sequencing • • Mortazavi A, Williams B A, Mc. Cue K, et al. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods, 2008, 5(7): 621— 628 Sugarbaker D J, Richards W G, Gordon G J, et al. Transcriptome sequencing of malignant pleural mesothelioma tumors. Proc Natl Acad Sci USA, 2008, 105(9): 3521— 3526 Digital expression profiling • • Ruby J G, Jan C, Player C, et al. Large-scale sequencing reveals 21 U-RNAs and additional micro. RNAs and endogenous si. RNAs in C. elegans. Cell, 2006, 127(6): 1193— 1207 Morin R D, O'Connor M D, Griffith M, et al. Application of massively parallel sequencing to micro. RNA profiling and discovery in human embryonic stem cells. Genome Res, 2008, 18(4): 610— 621 Ch. IP-seq • • Johnson D S, Mortazavi A, Myers R M, et al. Genome-wide mapping of in vivo protein-DNA interactions. Science, 2007, 316(5830): 1497— 1502 Robertson G, Hirst M, Bainbridge M, et al. Genome-wide profiles of STAT 1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods, 2007, 4(8): 651— 657


- Slides: 29