Nephrotic and Nephritic Syndrome in Children NITMED TUTORIALS







, hematuria")




 antedate glomerular")





















 Episodic hematuria associated with deposition")






 Sometimes called mesangiocapillary glomerulonephritis or lobar glomerulonephritis")


 Low serum C 3 and a dense thickening")











 reliably predict the severity")







: e. g. , Finnish-type congenital nephrotic syndrome")







 Causes 70 to 90% of")
: Histology Light microscopy: No obvious glomerular lesion")
: Clinical Consequences: 1. Edema is the most")
 3. Increase Susceptibility to Infection")

: Lab Findings: Manifestations of nephrotic syndrome Elevated")
: Treatment Hypercholesterolemia: The use of Lipid-lowering drugs")






 are recommended as initial therapy for children with steroid-resistant")















 include: • * Proteinuria")











- Slides: 115
Nephrotic, and Nephritic Syndrome in Children © NITMED TUTORIALS
Introduction Patients with glomerular disease usually have some hematuria with varying degrees of proteinuria. The diagnosis of glomerular injury can be delayed because patients will not realize they have microscopic hematuria (3 to 5 RBC/HPF), and only rarely with the exception of Ig. A Nephropathy and Sickle Cell Disease is Gross Hematuria present.
Microscopic hematuria may appear with anatomic lesions, interstitial nephritis, papillary necrosis, hypercalciuria, renal stones, cystic kidney diseases, or renal vascular injury. However, when red blood casts or dysmorphic red blood cells are found in the urine sediment, glomerulonephritis is likely.
Sustained proteinuria >1 – 2 g/24 hr is also commonly associated with glomerular disease. Benign proteinuria, or functional or transient proteinuria (<1 g/24 hr) is associated with fever, exercise, obesity, sleep apnea, emotional stress, and congestive heart failure.
Isolated proteinuria sustained over multiple visits is found in diabetic nephropathy, minimal change disease, mesangioproliferative glomerulonephritis, and FSGS.
Proteinuria in most adults with glomerular disease is nonselective, containing albumin and a mixture of other serum proteins, while in children with nil lesion from minimal change disease, the proteinuria is selective and composed largely of albumin.
Some patients with inflammatory glomerular disease, such as acute poststreptococcal glomerulonephritis or membranoproliferative glomerulonephritis (MPGN), have pyuria characterized by the presence of considerable numbers of leukocytes. This latter finding has to be distinguished from urine infected with bacteria.
Acute nephritic syndrome produces mild to moderate proteinuria (1 – 2 g/24 hr), hematuria with red blood cell casts, pyuria, hypertension, fluid retention, and a rise in serum creatinine associated with a reduction in glomerular filtration. Nephrotic syndrome produces heavy proteinuria (>3. 5 g/24 hr or a Urine protein : creatinine ratio >2 mg), hypertension, hypercholesterolemia (Cholesterol >200 mg/d. L), hypoalbuminemia (< OR = 2. 5 g/d. L), edema/anasarca, and microscopic hematuria.
While nephrotic-range proteinuria in adults is characterized by protein excretion of 3. 5 g or more per day, in children it is defined as protein excretion of more than 40 mg/m 2/hour or a first-morning urine protein/creatinine ratio of > OR + 2 to 3 mg.
Acute Nephritic Syndrome A. Poststreptococcal Glomeruolonephritis B. Subacute Bacterial Endocarditis C. Lupus Nephritis D. Goodpasture’s syndrome E. Ig. A Nephropathy (Berger Disease) F. Membranoproliferative Glomerulonephritis G. Membranous Glomerulonephritis
Acute Nephritic Syndrome H. Alport Syndrome I. Sickle Cell Disease J. Hemolytic Uremic Syndrome
Acute Nephritic Syndrome A. Poststreptococcal Glomeruolonephritis Acute poststreptococcal glomerulonephritis in underdeveloped countries usually affects children between the ages of 5 and 12 years (uncommon before the age of 3 yrs), but in developed countries is more typical in the elderly, especially in association with debilitating conditions. It is more common in males, and the familial or cohabitant incidence is as high as 40%
Skin and throat infections with particular M types of streptococci (nephritogenic strains) antedate glomerular disease; M types 47, 49, 55, 2, 60 and 57 are seen following impetigo and M types 1, 2, 4, 3, 25, 49, and 12 with pharyngitis. However most APSGN are sporadic Poststreptococcal glomerulonephritis due to impetigo develops 2 to 6 weeks after skin infection and 1 to 3 weeks after streptococcal pharyngitis.
Patients are at risk for developing encephalopathy and/or heart failure secondary to hypertension or hypervolemia. Hypertensive encephalopathy must be considered in patients with blurred vision, severe headaches, altered mental status, or new seizures. Respiratory distress, orthopnea, and cough may be symptoms of pulmonary edema and heart failure. Peripheral edema is common; NS develops is a minority (<5%) of childhood cases. Nonspecific symptoms such as malaise, lethargy, abdominal or flank pain are common.
The acute phase generally resolves within 6 to 8 weeks. Although urinary protein excretion and hypertension usually normalizes by 4 to 6 weeks after onset, persistent microscopic hematuria can persist for 1 to 2 year after the initial presentation.
Pathogenesis of ASPGN: Molecular mimicry whereby circulating antibodies elicited by streptococcal antigens react with normal glomerular antigens, in-situ immune complex formation of antistreptococcal antibodies with glomerular deposited antigen, and complement activation by directly deposited streptococcal antigens continue to be considered as probable mechanisms of immunologic injury.
The renal biopsy in poststreptococcal glomerulonephritis demonstrates hypercellularity of mesangial and endothelial cells, glomerular infiltrates of polymorphonuclear leukocytes, granular subendothelial immune deposits of Ig. G, Ig. M, C 3, C 4, and C 5 – 9, and subepithelial deposits (which appear as “humps”). Immonuflurescence microscopy reveals a pattern of “lumpy-bumpy” deposits of immunoglobulin and complement on the GBM and in the mesangium. Electron microscopy reveals electron-dense deposits “humps” of the GBM.
Five percent of children and 20% of adults have proteinuria in the nephrotic range. In the first week of symptoms, 90% of patients will have a depressed CH 50 and decreased levels of C 3 and normal levels of C 4. the serum C 3 level is significantly reduced in >90% of patients in the acute phase, and returns to normal 6 to 8 weeks after onset.
Confirmation of the diagnosis requires clear evidence of prior streptococcal infection. Rising ASO titer for poststreptococcal pharyngitis but rarely increases for steptococcal skin infection Antideoxyribonuclease B level is best to detect cutaneous streptococcal infection. Streptozyme screen measure multiple antibodies to different streptococcal antigens. Serologic evidence for streptococcal infections is more sensitive than the history of recent infections and far more sensitive than positive bacterial cultures obtained at the time of onset of acute nephritis
Indications for renal biopsy: the presence of acute renal failure, NS, absence of evidence of streptococcal infection, or normal complement levels, hematuria and proteinuria, diminished renal function, and/or low C 3 level that persist for more than 2 months after onset.
Five percent of children and 20% of adults have proteinuria in the nephrotic range. In the first week of symptoms, 90% of patients will have a depressed CH 50 and decreased levels of C 3 and normal levels of C 4. The serum C 3 level is significantly reduced in >90% of patients in the acute phase, and returns to normal 6 to 8 weeks after onset Positive cultures for streptococcal infection are inconsistently present (10 – 70%), but increased titers of ASO (30%), anti-DNAse (70%), or antihyaluronidase (40%) antibodies can help confirm the diagnosis. Consequently, the diagnosis of poststreptococcal glomerulonephritis rarale requires a renal biopsy.
Complications of APSGN: HTN, which can lead to hypertensive encephalopthy, acute kidney injury, heart failure, electrolyte imbalance (hyperkalemia, hyperphosphatemia, hypocalcemia, acidosis), seizures, and uremia. Early systemic antibiotic therapy for streptococcal throat and skin infections does not eliminate the risk of glomerulonephritis
Antibiotic treatment for streptococcal infection should be given to all patients and their cohabitants. There is no role of immunosuppressive therapy, even in the setting of crescents. Complete resolution of the hematuria and proteinuria in the majority of children occurs within 3 to 6 weeks of the onset of nephritis but 3 to 10% of children may have persistent microscopic hematuria, nonnephrotic proteinuria, or hypertension. Sodium restriction in diet, diuresis (usually with intravenous furosemide), and pharmacotherapy with calcium channel blockers, vasodilators, or ACEIs, are standard therapies used to treat hypertension.
Complete recovery occurs in >95% of children with APSGN.
Acute Nephritic Syndrome B. Subacute Bacterial Endocarditis Right-sided endocarditis untreated for a long time Blood cultures often negative Glomerulonephritis is unusual in acute bacterial endocarditis because it takes 10 to 14 days to develop immune complex-mediated injury, by which time the patient has been treated, often with emergent surgery.
Grossly, the kidneys in SBE have subcapsular hemorrhages with a “fleabitten” appearance, and microscopy on renal biopsy reveals focal proliferation around foci of necrosis associated with abundant mesangial, subendothelial, and subepithelial immune deposits of Ig. G, Ig. M, and C 3. The pathogenesis hinges on the renal deposition of circulating immune complexes in the kidney with complement activation. Primary treatment is eradication of the infection with 4 to 6 weeks of antibiotics, and if accomplished expeditiously, the prognosis for renal recovery is good.
Acute Nephritic Syndrome C. Lupus Nephritis Most severe in African-American female adolescents (The majority of children with SLE are adolescent females. The most common clinical sign of renal disease is proteinuria, but hematuria, hypertension, varying degrees of renal failure, and active urine sediment with red blood cell casts can all be present. Renal disease in childhood SLE is present in up to 80% of patients and is more active than that seen in adults. Occasionally, renal disease is the only presenting clinical manifestation.
The extrarenal manifestations of lupus are important in establishing a firm diagnosis of lupus because, while serologic abnormalities are common in lupus nephritis, they are not diagnostic. Lupus nephritis affects most pediatric patients, and although commonly presenting within the first year of diagnosis, may occur at any time during the course of disease. Patients with class V nephritis commonly present with nephrotic syndrome.
Anti-ds. DNA antibodies that fix complement correlate best with the presence of renal disease (The diagnosis of SLE is confirmed by the detection of circulating antinuclear antibodies and by demonstrating antibodies that react with native double-stranded DNA. Renal biopsy is the only reliable method of identifying the morphologic variants of lupus nephritis: Class 1 to 6. Transformation of the histologic lesions of lupus nephritis from one class to another is common
Remission is defined as a return to near-normal renal function and proteinuria < OR = 330 mg/d. L/day. Both Class 1 and 2 lesions are typically associated with minimal renal manifestation and normal renal function; nephrotic syndrome is rare. Patients with lesions limited to the renal mesangium have an excellent prognosis and generally do not need therapy for their lupus nephritis. Class 3 to 4 lesions are treated with high-dose steroids and either cyclophosphamide or mycophenolate mofetil for 2 to 6 months, followed by maintenance therapy with lower doses of steroids and mycophenolate mofetil
Therapy is initiated in all patients with prednisone at a dose of 1 – 2 mg/Kg/day in divided doses followed by a slow steroid taper over 4 to 6 months beginning 4 to 6 weeks after achieving remission. Nephrologist tend to avoid prolonged use of cyclophosphamide in patients of childbearing age without first banking eggs or sperm.
Patients with lupus nephritis class V, like patients with Idiopathic Membranous Nephropathy, are predisposed to renal-vein thrombosis and other thrombotic complications.
As a group approximately 20% of patients with lupus nephritis will reach end-stage disease, requiring dialysis or transplantation. Systemic lupus tends to be quiescent once there is renal failure, perhaps due to the immunosuppressant effects of uremia. Renal transplantation in renal failure from lupus, usually performed after approximately 6 months of inactive disease, results in allograft survival rates comparable to patients transplanted for other reasons.
Acute Nephritic Syndrome D. Ig. A Nephropathy (Berger Disease) Episodic hematuria associated with deposition of Ig. A in the mesangium. There is a male preponderance, a peak incidence in the 2 nd and 3 rd decades of life, and rare familial clustering. Ig. A nephropathy is predominantly a sporadic disease. There are geographic differences in the prevalence of Ig. A nephropathy, with 30% prevalence along the Asian and Pacific Rim and 20% in Southern Europe, compared to a much lower prevalence in northern Europe and North America.
No single causative gene has been identified. The clinical presentation of Ig. A nephropathy is often benign in comparison to that of adults. Clinical and laboratory evidence suggests close similarities between Henoch – Schonlein purpura (HSP) and Ig. A nephropathy HSP is distinguished clinically from Berger disease by prominent systemic symptoms, a younger age (< 20 years old), preceding infection, and abdominal complaints
HSP is the most common small vessel vasculitis in childhood that is characterized by a purpuric rash and commonly accompanied by arthritis and abdominal pain. The nephritis associated with HSP usually follows the onset of the rash, often presenting weeks to months after the onset of the rash. The severity of the systemic manifestations does not correlate with the severity of the nephritis. The prognosis of HSP nephritis for most patients is excellent.
Ig. A Nephropathy: Gross hematuria often occurs within 1 to 2 days of onset of an URTI or GIT infection, in contrast to the longer latency period observed in acute postinfectious glomerulonephritis. Normal serum levels of C 3 in Ig. A nephropathy help to distinguish this disorder from poststreptococcal glomerulonephritis.
Ig. A nephropathy is an immune-complex-mediated glomerulonephritis defined by the presence of diffuse mesangial Ig. A deposits often associated with mesangial hypercellularity. Ig. M, Ig. G, C 3, or immunoglobulin light chains may be co-distributed with Ig. A. Although the immunofluorescent pattern of Ig. A on renal biopsy defines Ig. A nephropathy in the proper clinical context, a variety of histologic lesions may be seen on light microscopy, including DPGN, segmental sclerosis; and, rarely, segmental necrosis with cellular crescent formation, which typically present as RPGN.
The two most common presentations of Ig. A nephropathy are recurrent episodes of macroscopic hematuria during or immediately following an upper respiratory infection often accompanied by proteinuria or persistent asymptomatic microscopic hematuria. Nephrotic syndrome, however, is uncommon. Proteinuria can also first appear late in the course of the disease. Rarely patients present with acute renal failure and a rapidly progressive clinical picture. Ig. A nephropathy is a benign disease for the majority of patients, and 5 – 30% of patients may go into remission, with other having hematuria but well preserved renal function.
ACEI in patients with proteinuria or declining renal function. Tonsillectomy, steroid therapy, and fish oil have all been suggested in small studies to benefit select patients with Ig. A nephropathy. When presenting as RPGN, patients typically receive steroids, cytotoxic agents, and plasmapheresis.
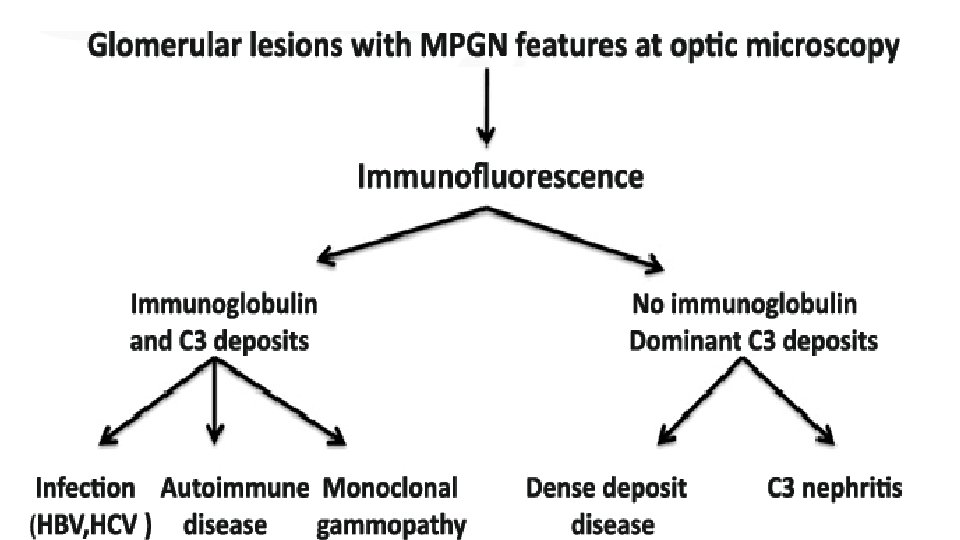
Acute Nephritic Syndrome F. Membranoproliferative Glomerulonephritis (MPGN) Sometimes called mesangiocapillary glomerulonephritis or lobar glomerulonephritis It is an immune-mediated glomerulonephritis characterized by thickening of the GBM with mesangioproliferative changes; 70% of patients have hypocomplementemia. Majority of cases of MPGN have low C 3 complement levels. MPGN is rare in African Americans, and idiopathic disease usually presents in childhood or young adulthood. MPGN is most common in the 2 nd decade of life.
MPGN is subdivided pathologically into type 1, type 2, and type 3 disease. Type 1 MPGN (Most Common): Hepatitis C infections, autoimmune diseases like lupus (SLE) or cryoglobulinemia, or Hepatitis B, neoplastic diseases (lung, breast, and ovary). Type 2 MPGN (Dense Deposit Disease): Usually idiopathic, except in patients with Complement Factor H deficiency, in the presence of C 3 nephritic factor and/or in partial lipodystrophy Type 3 MPGN: Usually idiopathic, except in patients complement receptor deficiency
Type 1 MPGN Mesangial proliferation with lobular segmentation Mesangial interposition between the capillary basement membrane and endothelial cells, producing a double-contour (tram-tracking) Subendotheial deposit with low serum C 3 are typical, although 50% of patients have normal levels of C 3 and occasional intramesangial deposits.
Type 2 MPGN (Dense Deposit Disease) Low serum C 3 and a dense thickening of the GBM containing ribbons of dense deposits. In type 2 MPGN, C 3 immunofluorescence typically is prominent without concomitant immunoglobulin.
Type 1 MPGN is secondary to glomerular deposition of circulating immune complexes or their in situ formation. Type 2 and 3 MPGN may be related to “nephritic factors, ” that stabilize C 3 Convertase and allow it to activate serum C 3. In contrast to MPGN where C 3 levels remain persistently low, C 3 returns to normal within 2 months after onset of postinfectious glomerulonephritis.
There is some evidence supporting the efficacy of treatment of primary MPGN with steroids, particularly in children, as well as reports of efficacy with plasma exchange and other immunosuppressive drugs. In secondary MPGN, treating the associated infection, autoimmune disease, or neoplasms is of demonstrated benefit. In particular, pegylated interferon and ribavirin are useful in reducing viral load.
Acute Nephritic Syndrome G. Membranous Glomerulonephritis Accounts for approximately 30% of cases of nephrotic syndrome in adults, with a peak incidence between the ages of 30 and 50 years and a male to female ratio of 2 : 1. MGN is rare in childhood and the most common cause of nephrotic syndrome in the elderly
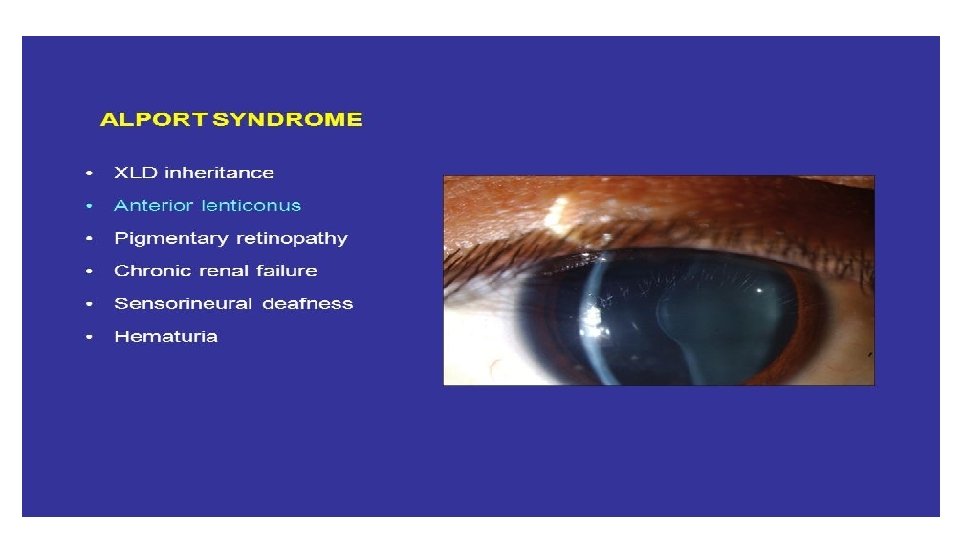
Acute Nephritic Syndrome • H. Alport Syndrome Patients with Alport Syndrome develop hematuria, thining and splitting of the GBM, mild proteinuria (<1 – 2 g/24 Hr), which appears late in the course, followed by chronic glomerulosclerosis leading to renal failure in association with bilateral sensorineural deafness. Some patients develop lenticonus of the anterior lens capsule (extrusion of the central portion of the lens into the anterior chamber) , “dot and flack” retinopathy, and rarely, mental retardation or leiomyomatosis.
Acute Nephritic Syndrome H. Alport Syndrome Approximately 85% of patients with Alport syndrome have an X-linked inheritance of mutations in the COL 4 A 5 gene (on chromosome Xq 2224) encoding the alpha 5 (IV) collagen chain of type IV collagen. Fifteen percent of patients have autosomal recessive disease from inheritance of mutations in the COL 3 A 5 or COL 4 A 5 gene (on chromosome 2 q 35 -37) encoding the alpha 3 (IV) or alpha 4 (IV) collagen chains of type IV collagen. Alport syndrome from mutations in the genes encoding for the alpha 3, alpha 4, alpha 5 chains of type 4 collagen, produces split-basement membranes with glomerulosclerosis
Clinical evaluation should include a careful eye examination and hearing tests. However, the absence of extrarenal symptoms does not rule out the diagnosis More likely to end with ESRD than Ig. A nephropathy
Acute Nephritic Syndrome I. Sickle Cell Disease Although individuals with Hb. AS are usually asymptomatic, most will gradually develop hyposthenuria due to subclinical infarction of the renal medulla, thus predisposing them to volume depletion. In Hb. SS patients, vessels occlusions in the kidney produce glomerular hypertension, FSGS, interstitial nephritis, and renal infarction with hyposthenuria, microscopic hematuria, and even gross hematuria; some patients also present with MPGN.
By the 2 nd or 3 rd decade of life, persistent vasoocclusive disease in the kidney leads to varying degrees of renal failure, and some patients end up on dialysis. Treatment is directed to reducing the frequency of painful crises and administering ACEI in the hope of delaying a progressive decline in renal function.
Acute Nephritic Syndrome J. Hemolytic Uremic Syndrome TTP and HUS constitute a spectrum of thrombotic microangiopathies with a general feature of idiopathic thrombocytopenic purpura, hemolytic anemia, fever, renal failure, and neurologic disturbances. When patients, particularly children, have more evidence of renal injury, their condition tend to be called HUS.
HUS is most common in preschool and school-age children, but it can occur in adolescents and adults. In HUS caused by toxigenic E. coli, onset of HUS occurs a few days after onset of gastroenteritis with fever, vomiting, abdominal pain, and diarrhea. The prodromal intestinal symptoms may be mild to severe requiring hospitalization. The diarrhea is often bloody, but necessarily so. Following the prodromal illness, the sudden onset of pallor, irritability, weakness, and lethargy coincides with the onset of HUS.
Oliguria can be present in early stages but may be masked by ongoing diarrhea, because the prodromal enteritis often overlaps the onset of HUS. Thus patients with HUS can present with either significant dehydration from the enteritis or volume overload from the renal insufficiency depending on which predominates, and the amount of fluid that has been administered.
No presenting features (leukocytosis, severe prodromal enteritis, hyponatremia, antibiotic use) reliably predict the severity of HUS in any given patient. Mild renal insufficiency at presentation can rapidly evolve into oliguric or anuric renal failure. The combination of rapidly developing renal failure and severe hemolysis can result in lifethreatening hyperkalemia. Volume overload, hypertension, and severe anemia can all develop soon after onset of HUS, and together can precipitate heart failure.
In each form of HUS, capillary and endothelial injury in the kidney leads to localized thrombosis, particularly in the glomeruli, causing a direct decrease in GFR. The diagnosis is made by the combination of microangipathic hemolytic anemia with schistocytes, thrombocytopenia, and some degree of renal involvement.
The majority of patients with HUS have some CNS involvement. Seizures and significant encephalopathy are the most common manifestations in those with severe CNS involvement, resulting from focal ischemia secondary to microvascular CNS thrombosis.
On examination of the kidney tissue there is evidence of glomerular capillary endotheliosis associated with platelet thrombi, damage to the capillary wall, and formation of fibrin material in and around the glomeruli. TTP/HUS is also seen in pregnancy, oral contraceptives or quinine, calcineurin inhibitors, antiplatetel agents (ticlopidine and clopidogrel), or following HIV infection
Childhood HUS is associated with enterohemorrhagic diarrhea. Childhood HUS is caused by shiga-like toxin released by E. coli (STEC) 0157: H 7, 0104: H 4 (Western countries) and occasionally by toxins of Shigella dysenteriae type 1 (Asia and southern Africa). The reservoir of STEC is the intestinal tract of domestic animals, usually cows. Patients with childhood HUS from infectious diarrhea are not given antibiotics.
Platelets should generally not be administered , regardless of platelet count, to patients with HUS because they are rapidly consumed by the active coagulation and theoretically can worsen the clinical course. Despite low platelet counts, serious bleeding is very rare in patients with HUS.
# The following about HUS is correct It is the most common cause of ARF in young children Classically is characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia, and uremia It is also associated with shigella It is associated with significant leukocytosis
2. Nephrotic Syndrome A. Idiopathic Nephrotic Syndrome (Minimal Change Disease (Approximately 85% of total cases of nephrotic syndrome in children), FSGS, MN, MPGN, Ig. A Nephropathy)
B. Genetic Disorders: i. Nephrotic Syndrome (Typical): e. g. , Finnish-type congenital nephrotic syndrome (absence of nephrin), Denys-Drash syndrome) Ii. Proteinuria with or without Nephrotic Syndrome (e. g. Nail-patella syndrome, Alport syndrome) Iii. Multisystem syndromes with or without Nephrotic Syndrome (e. g. , Charcot-Marie-Tooth disease) Iv. Metabolic Disorders with or without Nephrotic Syndrome (e. g. , Alagille syndrome, alpha 1 -antitrypsin deficiency, glycogen storage disease, sickle cell disease)
Congenital Nephrotic Syndrome: A number of hereditary proteinuria syndromes are caused by mutations in genes that encode critical protein components of the glomerular filtration apparatus. Congenital NS from mutations in NPHS 1 (nephrin) and NPHS 2 (podocin) affect the slit-pore membrane at birth.
Congenital Nephrotic syndrome is defined as NS manifesting at birth or within the 1 st 3 months of life. Congenital NS may be classified as primary or as secondary to a number of etiologies such as in utero infections (cytomegalovirus, toxoplasmosis, syphilis, HBV, HCV, HIV), infantile SLE, or mercury exposure.
C. Secondary Causes of Nephrotic Syndrome: i. Infections (Endocarditis, HBV, HCV, HIV-1, Infectious Mononucleosis, malaria, toxoplasmosis, schistosomiasis) Ii. Drugs (e. g. , captopril, pecillamine, Gold, NSAIDs, pamidronate, interferon, mercury, heroin, lithium) Iii. Immunologic or allergic disorders (Vasculitis syndrome [SLE, HSP], Beesting, food allergies, serum sickness) Iv. Malignancy (lymphoma, leukemia, solid tumors)
Secondary nephrotic syndrome should be suspected in patients > 8 years and those with hypertension, hematuria, renal dysfunction, extrarenal symptoms (rash, arthralgias, fever), or depressed serum complement levels. In certain areas of the world, malaria and schistosomiasis are the leading causes of nephrotic syndrome.
2. Nephrotic Syndrome Nephrotic syndrome affects 1 – 3 per 100, 000 children < 16 years of age. Without treatment, nephrotic syndrome in children is associated with a high risk of death, most commonly from infections. Fortunately, 80% of children with nephrotic syndrome respond to corticosteroid therapy.
Most children with nephrotic syndrome have a form of primary or idiopathic nephrotic syndrome (Table 527 -1). Glomerular lesions associated with idiopathic nephrotic syndrome include minimal change disease (the most common), FSGS, MPGN, C 3 glomerulopathy, and membranous nephropathy. Nephrotic Syndrome may also be secondary to systemic diseases such as SLE, HSP, malignancy (lymphoma and leukemia), and infections (hepatitis, HIV, and malaria).
Congenital Nephrotic Syndrome: A number of hereditary proteinuria syndromes are caused by mutations in genes that encode critical protein components of the glomerular filtration apparatus.
2. Nephrotic Syndrome A. Minimal Change Disease (nil lesion) Causes 70 to 90% of nephrotic syndrome in childhood but only 10 – 15% of nephrotic syndrome in adults MCD usually presents as a primary renal disease but can be associated with several other conditions, including Hodgkin’s disease, allergies, or use of NSAIDs MCD : 2 to 6 years; M: F = 2 : 1
MCD A. Minimal Change Disease (nil lesion): Histology Light microscopy: No obvious glomerular lesion Immuniflurescent miscroscopy: Negative for deposits in the mesangium , or occasionally shows small amounts of Ig. M in the mesangium Electron microscopy: Effacement of the foot process supporting the epithelial podocytes with weakening of slit-pore membrane.
MCD A. Minimal Change Disease (nil lesion): Clinical Consequences: 1. Edema is the most common presenting symptom Underfill hypothesis (a fall in plasma protein with a reduction in oncotic pressure and intravascular fluid depletion (low BP, tachycardia, and elevated hemoconcentration) Overfill hypothesis (primary sodium retention with subsequent volume expansion and leakage of excess fluid into the interstitium)
2. Hyperlipidemia (increase in cholesterol, triglycerides, LDL, and VLDL) 3. Increase Susceptibility to Infection (e. g. , cellulitis, spontaneous bacterial peritonitis, and bacteremia) especially from encapsulated bacteria and in particular pneumococcal disease Spontaneous bacterial peritonitis presents with fever, abdominal pain, an peritoneal signs. Although pneumococcus is the most frequent cause of peritonitis, gram-negative bacteria als are associated with a significant number cases.
4. Hypercoagulabilty. There is an increase in hepatic production of fibrinogen along with urinary losses of antithrombotic III and protein S. Deep Venous Thrombosis may occur in nay venous bed, including the cerebral venous sinus, renal vein, and pulmonary veins. Important features of MCD are the absence of hypertension and gro hematuria.
MCD A. Minimal Change Disease (nil lesion): Lab Findings: Manifestations of nephrotic syndrome Elevated BUN in 15 – 30% Normal Complement Levels
MCD A. Minimal Change Disease (nil lesion): Treatment Hypercholesterolemia: The use of Lipid-lowering drugs is uncommon in children, low-fat diet Edema secondary to salt and water retention: Judicious use of diuretics, sodium restriction (<1500 mg daily), and cautious use of intravenous albumin infusions, if indicated) Hypercoagulable state: anticoagulants (heparin, LMW heparin, and warfarin) Proteinuria: ACEI
MCD In children, the abnormal urine principally contains albumin with minimal amounts of higher-molecular-weight proteins, and is sometimes called selective proteinuria.
MCD Although up to 30% of children have a spontaneous remission, all children today are treated with dsteroids; only children who are nonresponders are biopsied in this setting. Primary Responders: Patients who have complete remission (<0. 2 mg/24 hr proteinuria) after a single course of prednisone Steroid Dependent: Patients relapse as their steroid dose is tapered Frequent Relapsers: Two or more relapses in the 6 months following taper Steroid Resistant: Patients fail to respond to steroid therapy
Approximately 80 to 90% of children respond to steroid therapy. Response is defined as the attainment of remission within the initial 4 weeks of corticosteroid therapy. Remission consists of a urine protein : creatinine ratio of <0. 2 or <1+ protein on urine dipstick for 3 consecutive days Relapse of nephrotic syndrome is defined as a urine protein: creatinine ratio of >2 or > or = 3+ protein on urine dipstick testing for 3 consecutive days
Relapses are common especially in younger children, and are often triggered by upper respiratory or gastrointestinal infections. Children are classified as infrequent relapsers or frequent relapse, and as being steroid dependent based on the number of relapses in a 12 month period or their inability to remain in remission following discontinuation of steroid therapy. Steroid resistance is defined as the failure to achieve remission after 8 weeks of corticosteroid therapy. Steroid – Resistant NS is usually caused by FSGS, or membranoproliferative glomerulonephritis.
Ninety to 95% of children will develop a complete remission after 8 weeks of steroid therapy Patients with steroid resistance may have FSGS. Relapses occur in 70 to 75% of children after the first remission, and early relapse predicts multiple subsequent relapses. The frequency of relapses decreases after puberty, although there is an increased risk of relapse following the rapid tapering of steroids in all age groups.
Prednisone is first-line therapy, either given daily or on alternate days. Other immunosuppressive drugs, such as cyclophosphamide, chlorambucil, and mycophenolate mofetil, are saved for frequent relapsers, steroid-dependent, or steroid-resistant patients. Cyclosporine can induce remission, but relapse is also common when cyclosporine is withdrawn.
Calcineurin inhibitors (cyclosporine or tacrolimus) are recommended as initial therapy for children with steroid-resistant NS. Children should be monitored for side effects, includinh hypertension, nephrotoxicity, hirsutism, and gingival hyperplasia. Steroid-dependent patients, frequent relapsers, and steroid-resistant patients are candidaytes for alternative therapies, particularly if they have severe corticosteroid toxicity (cushingoid appearance, hypertension, cataracts, and/or growth failure).
In children with presumed MCNS, prednisone or prednisolone should be administered as a single daily dose of 60 mg/m 2/day or 2 mg/Kg/day to a maximum of 60 mg daily for 4 to 6 weeks followed by alternate-day prednisone (starting at 40 mg/m 2 qod or 1. 5 mg/Kg qod) for a period ranging from 8 weeks to 5 months, with tapering of the dose. ACEI and Angiotensin II receptor blockers may be helpful as adjunct therapy to reduce proteinuria in steroid-resistant patients.
The diagnosis of nephrotic syndrome is confirmed by urinalysis with first morning urine protein : creatinine ratio and serum electrolytes, BUN, creatinine, albumin, an cholesterol levels; evaluation to rule out secondary forms of NS (children >10 yr): Complement C 3 level, antinuclear antibody, ds-DNA, and HBV and HCV. And HIV in high-risk populations; kidney biopsy (for children > or = 12 yr, who are less likely to have MCNS)
The differential diagnosis of the child with marked edema includes protein losing enteropathy, hepatic failure, heart failure, acute or chronic glomerulonephritis, and protein malnutrition. A diagnosis other than MCNS should be considered in children <1 yr of age, with a positive family history of NS, and/or the presence of extrarenal findings (e. g. , arthritis, rash, anemia), hypertension or pulmonary edema, acute or chronic renal insufficiency, and gross hematuria.
Cyclophosphamide prolongs the duration of remission and reduces the number of relapses in children with frequently relapsing and steroiddependent nephrotic syndrome. The potential side effects of the drug (neutropenia, disseminated vasculitis, hemorrhagic cystitis, alopecia, sterility, increased risk of future malignancy) should be carefully reviewed with the family before initiating treatment.
Tuberculosis must be ruled out prior to starting immunosuppressive therapy with corticosteroids. Children with features that make MCNS less likely (gross hematuria, hypertension, renal insufficiency, hypocomplementemia, or age <1 yr or > 12 yr) should be considered for renal biopsy before treatment.
Glucocorticoids may increase the body mass index in children who are overweight when steroid therapy is initiated, and these children are more likely to remain overweight. Anticipatory dietary counseling is recommended.
• # Immunization for Children with Nephrotic Syndrome • * Give full pneumococcal vaccination (with the 13 -valent conjugate vaccine and 23 -valent polysaccharide vaccine) and influenza vaccine annually to the child and household contacts • * Differ vaccination with live vaccines until the prednisone dose is below either 1 mg/Kg daily or 2 mg/Kg on alternate days • * Give Varicella zoster immune globulin to NS children exposed to varicella and immunize healthy household contacts with live vaccines to minimize the risk of transfer to the immunosuppressed child
• Most children with steroid-responsive NS have repated relaspes, which generally decrease in frequency as the child grows older. • It is important to indicate to the family that the child with steroidresponsive NS is unlikely to develop chronic kidney disease, that the disease is rarely hereditary, and that the child (in the absence of prolonged cyclophosphamide therapy) will remain fertile.
• # In Minimal change nephrotic syndrome • * Peak incidence is in children below 2 years of age • * Females predominate • * Gross hematuria occurs • * C 4 levels are low • * Hypertension is a common feature
• # Heavy proteinuria in Nephrotic syndrome results in the following • * Increased tendency to thromboembolism • * Anasarca • * Increased risk of infection • * Hypertension • * Cardiac failure
• # Concerning acute post-streptococcal glomerulonephritis: • * Usually occurs in infants • * May present with convulsions • * Low C 3 levels occurs in the acute phase of illness • * Is caused by non-hemolytic streptococci in 40% of cases • * Treatment with ampicillin reduces the time course of illness
• # Long term steroid therapy in children with nephrotic syndrome could lead to: • * VZV infection • * Avascular necrosis of the head of the femur • * Cataract • * Adrenal suppression • * Stunting
• # Indications for renal biopsy in childhood nephrotic syndrome • * Renal failure not attributable to hypovolemia • * Age of onset less than 6 months • * Hematuria • * Hypertension • * Steroid resistance
• # The following definitions regarding nephrotic syndrome are correctly matched: • * Remission: urinary protein excretion 0 f <4 mg/m 2/hr or albustix = 0 – trace for 3 consecutive days • * Relapse: urinary protein excretion <40 mg/m 2/hr or albustix = 0/trace for 3 consecutive days, after previously being in remission • * Frequent relapses: two or more relapses within six months of initial response • * Steroid dependence: relapses occurring during steroid therapy or within 14 days of its cessation • * Steroid resistance: failure to achieve response in spite of 2 weeks on prednisolone 60 mg/m 2/day
• # The following features are present in diarrhea positive HUS: • * Elevated liver enzymes • * Raised serum creatinine level • * Thrombocytopenia • * Abnormal red cell morphology with the presence of helmet cells, spherocytes, schistocytes, and burr cells • * DIC
• # Characteristics of Berger’s disease (Ig. A nephritis) include: • * Proteinuria in 70% of cases • * Female preponderance • * Deafness • * End stage renal failure within a year • * Exacerbation by viral upper airway infection
• # Concerning Ig. A nephropathy: • * It is the commonest cause of ARF in the black population • * Condition does not require any treatment • * Intravenous cyclophosphamide may be required • * It is cause by an enterovirus • * Most children end up with ESRD
• # Concerning Ig. A nephropathy and HSP: • * They have the same aetiopathogenesis • * Inheritance is autosomal recessive • * Both of have typical generalized skin rashes • * Prednisolone may be used in both conditions • * Gastrointestinal manifestations are more profound in Ig. A
• # With regards to congenital nephrotic syndrome • * Manifests usually after first six months of life • * It is the same thing as infantile nephrotic syndrome • * Responds well to steroids • * Can be caused by maternal transmission of syphilis • * Most affected children end up with renal tuberculosis
• # The following features may be found in children with congenital nephrotic syndrome: • * Severe malnutrition • * Hypothyroidism • * Large placenta of 25% or more of the birth weight • * Ascites • * Large echogenic kidneys on ultrasound
• # In congenital NS: • * NSAIDs may be indicated to control proteinuria • * Increased mesangial matrix with glomerulosclerosis is typical renal histology • * Most children survive the first three years without renal replacement therapy • * Renal transplantation is treatment of choice • * May occur in families
• # In childhood nephrotic syndrome: • * Aggressive diuresis may result in AKI • * Every child should receive oral prednisolone for at least 6 to 8 weeks • * SLE may be responsible • * Prednisolone is indicated in those secondary to HIV infection • * Minimal change disease is never associated with hematuria
• # Fever and intense generalized abdominal pain in a child with NS may indicate: • * Severe malaria • * Pyelonephritis • * Bilateral basal pneumonia • * Spontaneous peritonitis • * Cystitis
• # In the management of childhood NS: • * Main indicator for ACEI use is for blood pressure control • * Oral aspirin may be indicated • * Renal biopsy is mandatory in children less than one year • * ACEI may reduce urinary protein loss • * Intravenous albumin can correct proteinuria
• # The following are histologic characteristics of minimal change NS: • * Markedly increased mesangial matrix and hypercellularity on light microscopy • * C 3/Ig. G glomerular basement membrane deposits • * No glomerular or tubular abnormalities on light microscopy • * Marked podocyte hyperplasia on light microscopy • * Fusion/effacement of podocytes on electron microscopy
• # Risk factors for thromboembolic phenomena in NS include: • * Hyperlipidemia • * Decreased platelet aggregation • * Increased procoagulatory cofactors • * Decreased protein C • * Decreased procoagulatory factors
• # Regarding Alports syndrome: • * Can only be inherited as an X-linked disorder • * It comprises renal disease, hearing defect, liver disease, and ocular abnormalities • * Hematuria is a prominent feature • * All of the above • * None of the above