Nemoci glomerul a tubul Pednka z patologick fyziologie

Nemoci glomerulů a tubulů Přednáška z patologické fyziologie 28. 3. 2007

Stavba glomerulu
 heterogenní skupina chorob Rozdělení: a) Primární glomerulopatie b) Sekundární glomerulopatie –")
Nemoci glomerulů (glomerulopatie) heterogenní skupina chorob Rozdělení: a) Primární glomerulopatie b) Sekundární glomerulopatie – jedním z projevů systémového, cévního, metabolického nebo genetického onemocnění postihujícího i jiné orgány Častým mechanizmem vzniku glomerulopatií imunopatologické mechanizmy

Hlavní mechanizmy glomerulárního poškození U neproliferativních glomerulopatií: § § poškození autoprotilátkou poškození zprostředkované komplementem U proliferativních glomerulopatií: § § poškození cirkulujícími zánětlivými buňkami (zejména neutrofily a makrofágy) poškození lokálně aktivovanými rezidentními (např. mezangiálními buňkami)

Imunopatologické procesy Poškození ledvin závisí: - na velikosti imunokomplexů na rychlosti jejich depozice do tkání

Klasifikace glomerulopatií n n n Klinická: primární x sekundární Dle časového průběhu: akutní x subakutní x chronické Dle nálezu v renální biopsii: fokální x segmentální x difúzní n Dle buněčnosti: neproliferativní x proliferativní n Dle imunofluorescence:

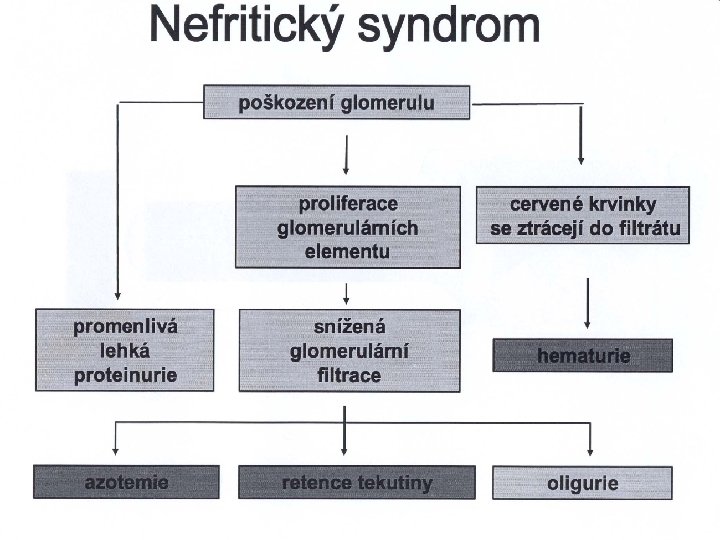
Důsledky a klinické projevy poruch glomerulů Poruchy funkce glomerulů – následky: n n n Pokles glomerulární filtrace event. zmenšení filtrační frakce Porucha separace bílkovin plazmy mezi intra- a extravaskulárním prostorem glomerulu Průnik krevních elementů do filtrátu Klinické projevy: n n glomerulární proteinurie glomerulární hematurie zvýšení azotémie hypervolémie a edémy, příp. hypertenze

Poruchy propustnosti glomerulární membrány n n Glomerulární membrána je vysoce permeabilní pro vodu (100 x vyšší) Průchodnost membrány v závislosti na velikosti molekuly určují zejména kolagenní struktury v bazální membráně (lamina densa – kolagen typu IV) n Elektrostatická repulsní bariéra (negat. nabité glykoproteiny s kys. sialovou – glykokalyx, glukozaminoglykany a hyaluronová kys. ) n Mezi výběžky podocytů je štěrbina přemostěná membránou – nefrin - vrozené poruchy (NS finského typu. . . ) - získané poruchy (DM)
 Funkční proteinurie")
Proteinurie - patologický symptom označující přítomnost proteinů v moči (nad fyziologické množství) Funkční proteinurie - příčina není v poruše funkce glomerulární membrány (ortostatická, pochodová, při horečce, po větší fyzické námaze…), ale ve změněných hemodynamických poměrech v glomerulech ( hydrostat. tlak v kapilárách) - charakter neselektivní proteinurie Proteinurie glomerulární - - selektivní: provází poškození glomerulární membrány, při kterých zůstává intaktní lamina densa – dojde ke ztrátě glykokalyxu z povrchu endotelií a podocytů – např. v důsledku zánětu (zadrženy větší proteiny než albumin) neselektivní: důsledkem většího strukturního poškození včetně lamina densa a hlavně podocytů a membrány mezi jejich výběžky (v moči „postalbuminová frakce proteinů)

n Patologické proteiny – mohou procházet do glomerulárního filtrátu ( -dimery globinu, myoglobin, lehké Ig řetězce či tzv. Bence Jonesova bílkovina) Tubulární proteinurie – nález proteinů v moči při intaktní funkci glomerulů a jejich membrány, ale při poškození tubulů - snížená zpětná resorpce malých plazmatických bílkovin (nejvyšší zastoupení 2 mikroglobulinu v moči) Proteinurii je třeba posuzovat i z kvantitativního hlediska. - citlivost běžných dg. metod je nastavena tak, aby odhalila klinicky závažnou proteinurii, při které se do moči dostává více než 0. 5 g proteinů.

Hematurie - značí zvýšenou přítomnost erytrocytů v moči. Příčiny: n krvácení z vývodných cest močových - v moči je velké množství Ery při malé nebo neprokazatelné proteinurii - barva jasně červená n průnik Ery přes glomerulární membránu - provázen měřitelnou proteinurií různého stupně - kouřové zbarvení v průhledu a rudě hnědá barva (Cola či čaj) v dopadajícím světle (v důsledku dlouhodobého vystavení Hb kyselému p. H moče) - deformity Ery mikroskopická x makroskopická

Výskyt válců v močovém sedimentu n n Jde o „odlitky“ určitých částí ledvinových tubulů, příp. s přichycenými zbytky buněk (ery, leu, epitelií) Matrix válců je tvořena Tammovým-Horsfallovým proteinem secernovaným do glomerulárního filtrátu tubulárními buňkami Základem je precipitace zahuštěného T-H proteinu s albuminem v prostředí kyselého p. H moče Zvýšení jeho výskytu v moč. sedimentu (Addisonův sediment, norm. do 105 za 24 hod) ukazuje zvýšený průnik albuminu glomerul. membránou (a tedy poškození glomerulů) - výskyt ery válců svědčí pro hematurii glomerulárního původu - epitelové a leukocytové válce – poškození tubulů a přítomnost zánětu v intersticiu - tukové válce – u nemocných s nefritickým syndromem a lipidurií

Typické syndromy Ø NEFRITICKÝ Ø NEFROTICKÝ Ø Chronická glomerulonefritis

Patogeneze nefritických nemocí
 § § Po infekčním onemocnění kůže nebo faryngu („nefritogenní“ kmeny")
Akutní glomerulonefritida (poststreptokoková GN) § § Po infekčním onemocnění kůže nebo faryngu („nefritogenní“ kmeny streptokoků s „patogenními“ Ag – např. endostreptosin) Pt proti strepto reagují křížově s vimentinem za tvorby imunokomplexů nefritis se rozvíjí po latentní periodě (2 -3 tý) klinický obraz: nefritického syndromu histologicky: endokapilární proliferace mezangiálních i endotelových buněk se subepitelovými („humps“) i subendotelovými depozity C 3, event. Ig. G Akutní difúzně proliferativní GN
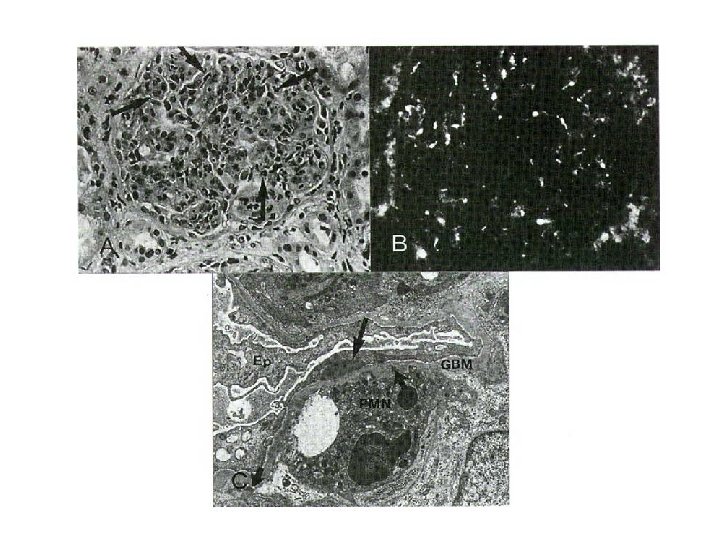

Postinfekční nonstreptokoková glomerulonefritida § Akutní glomerulonefritis se může vyskytnout i při jiných infekcích: - stafylokoky - pneumokoky - Klebsiella pneumonie - herpes virus - EBV - virus hepatitidy apod. § GN při infekční endokarditidě § GN při viscerálním abscesu (nejčastěji plicním) Histologický i klinický obraz – se podobá obrazu při poststreptokokové GN
 § § Heterogenní skupina nemocí, je charakterizována postižením většiny glomerulů")
Rychle progredující glomerulonefritidy (RPGN) § § Heterogenní skupina nemocí, je charakterizována postižením většiny glomerulů (> 50 -70% ) srpky s klinicky rychlou progresí do SL Vznik srpků: při poškození glomerulární kapilární stěny, které umožňuje průnik proteinů (fibrinogenu) a buněk (monocytů) do Bowmanova prostoru stimulace proliferace parietálních epiteliálních buněk depozita fibrinu utlačují kapiláry ( GFR a zánik glomerulu)
 proti bazální")
Tři typy RPGN § GN s tvorbou protilátek (Ig. G, Ig. A) proti bazální membráně (anti-GBM) - lineární depozita Ig (+ alveolokapilární síť) § Goodpastuerův sy GN s granulárními depozity Ig a komplementu - vznik srpků znamená komplikaci primárně endokapilárně proliferativní GN (u Ig. A nefropatie, SLE, akutní GN apod. ) § GN imunofluorescenčně negativní - ANCA (Pt proti cytoplazmě neutrofilních leukocytů) klinicky – s obrazem systémového onemocnění (Wegenerova granulomatóza) - izolovaného postižení ledvin (renálně limitovaná vaskulitis) Srpková GN

Goodpasterův syndrom § § § Charakterizované přítomností Pt proti bazální membráně glomerulů (a alveolokapilární membráně) Etiologie: kombinace exogenních faktorů (kouření, infekce, toxické látky) a geneticky vnímavého terénu (HLA B 7, DR 2) Patogeneze: GBM je tvořena kolagenem IV s navázanými proteiny (lamininem, entaktinem, tenascinem) a proteoglykany Goodpasterův antigen (lokalizován do C-terminální nekolagenní globulární domény (NC 1) molekuly 3 řetězce kolagenu IV tvorba Pt (Ig. G 1 schopné vázat komplement) poškození BM § Klinický obraz: GN + hemoptýza + těžká anémie (hypochromní mikrocytární)

Pomalu progredující glomerulonefritidy § § Skupina GN, které se označují jako membranoproliferativní GN 2 typy: u 1. typu jsou: - hladiny cirkulujícího komplementu - přítomny subendoteliální a mezangiální depozita klinicky: proteinurie nebo obraz nefrotického syndromu u 2. typu nalézáme: - aktivace komplementu je způsobená nefritickým faktorem C 3 - přítomna intramembránová depozita klinicky: proteinurie nebo obraz nefritického syndromu (jako u RPGN)
 § § § Zejména u dětí Patogeneze nejasná")
Glomerulopatie s minimálními změnami (lipoidní nefróza) § § § Zejména u dětí Patogeneze nejasná – souvislost s virovými infekcemi, imunizací, atopií, aplikací některých léků (antiflogistika) Výskyt spojený s některými antigeny HLA (DRw 7, B 8, B 12 …) Podstatou: ztráta negat. náboje ( permeabilita pro bílkoviny) Histologické změny: minimální (mírná proliferace mesangia, edematózní podocyty, splynutí („ztráta“) pedicel) § Léčba: kortikoidy
 glomeruloskleróza § vážnější stupeň předchozí formy - ložisková: < 50% glomerulů je")
Fokální (segmentální) glomeruloskleróza § vážnější stupeň předchozí formy - ložisková: < 50% glomerulů je poškozeno - difúzní: > 50% glomerulů je poškozeno - segmentální: poškozena jen část kapiláry glomerulu Glomeruloskleróza - obliterace lumen kapilár Je důsledkem – primárního poškození podocytů, v určitých segmentech (částech) glomerulů je zvýšená celularita

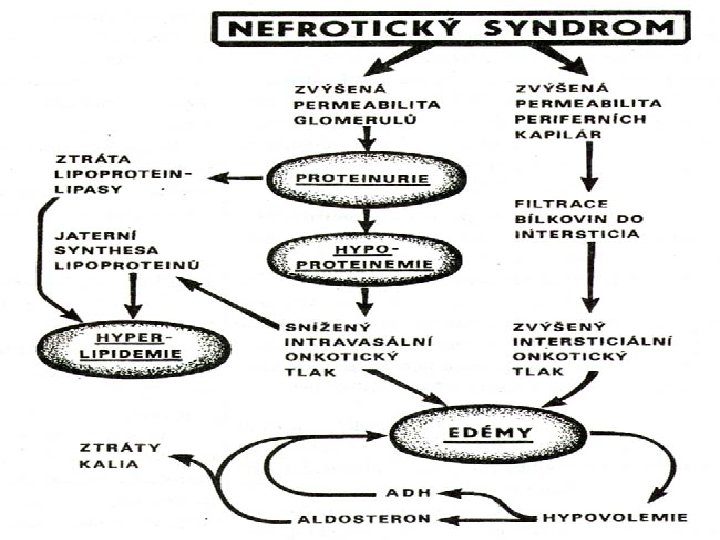
Membranózní glomerulopatie n n n Difúzní ztluštění glomerulární kapilární stěny, způsobené depozicí IK mezi podocyty a bazální membránu Silná vazba na HLA (B 8, DR 3) a geny alternativní cesty aktivace komplementu (Bf) Často sekundární etiologie: - léky (zlato, penicilamin…) - tumory (zejm. střeva) - infekce (hepatitis B) Klinický obraz: nefrotický syndrom s mikroskopickou hematurií a někdy hypertenzí Léčba: dle etiologie

Stadia membranózní GN

Idiopatická membranózní glomerulopatie
 glomerulopatie - je charakterizována klinicky nefrotickým syndromem Histologicky: zmnožení mesangia a jeho")
Membranoproliferativní (mezangiokapilární) glomerulopatie - je charakterizována klinicky nefrotickým syndromem Histologicky: zmnožení mesangia a jeho periferní expanze do stěny glom. kapilár s redukcí jejich průsvitu 2 typy: klasický – proliferace a zmnožení mezangiální matrix s šířením do stěny kapiláry mezi endotelie a bazální membránu nemoc denzních depozit – nerovnoměrné hromadění amorfního materiálu v lamina denza bazální membrány - - etiopatogeneze: - vazba na předchozí infekci (bakteriální endokarditis, abdominální absces, lepra…) - genetické faktory (HLA B 8, DR 3…) klinicky: nefrotická proteinurie s mikro hematurií, hypertenzí, anémií a hypokomplementémií ( C 3) - Prognóza: nepříznivá
 n n Mezangioproliferativní GN s depozity Ig. A, příp.")
Ig. A nefropatie (Bergerova choroba) n n Mezangioproliferativní GN s depozity Ig. A, příp. C 3 Příčina: - není známá, manifestace vázané na infekci (HCD)- latence 2 -3 dny - asociace s HLA (DQ, DP) ? ? Snad abnormální glykosylace Ig. A T-ly produkují množství IL-2 (+ IR-2 R) a jsou tedy trvale stimulovány nadprodukce polymerního Ig. A B-ly n Projevy: asymptomatická hematurie až nefrotický syndrom
 Charakterizována")
Chronická glomerulonefritis § § Společný konečný stav řady glomerulárních nemocí („end stage kidney“) Charakterizována proměnlivým rozsahem sklerotizace a proliferace Patogeneze: ztráta nefronů hyperperfúze hyperfiltrace skleróza glomerulů Stabilní nebo pokračuje do selhání ledvin

Glomerulopatie ve spojení se systémovými nemocemi Systémový lupus erythematodes § § § Nefritida se objevuje u 50 -70% nemocných se SLE (abnormality při biopsii vždy) Klinický obraz: - asi u ¼ klinické příznaky v době diagnózy - variabilní, modifikován základní terapií Histologické změny: WHO klasifikace – normální glomeruly (typ I) - mezangiální GN (typ II) - fokálně proliferativní GN (typ III) - difúzní proliferativní GF (typ IV) - membranózní GN (typ V) - glomerulání skleróza (typ VI)

Systémové vaskulitidy § § heterogenní skupina chorob charakterizovaná nekrotizujícím zánětem cév, který vede k poruše prokrvení oblasti zásobované příslušnými cévami Etiologie: primární x sekundární Patogeneze: - poškození IK - ANCA (pauciimunní typ) - poškození buňkami (IV. typ) Ledviny bývají postižené v závislosti na velikosti cév, které jsou vaskulitidou postiženy – postižení je značně variabilní

Henochova-Schönleinova purpura - systémová vaskulitida postihující cévy středního kalibru § zejména u dětí a mladších osob § často po infekci HCD, lécích § klinický obraz: - non-trombocytopenická purpura (příčinou je leukocytoklastická dermální vaskulitida) - postižení kloubů, serózních blan, GIT a glomerulů změny takřka identické s Ig. A nefropatií

Polyarteritis nodosa - § § § je onemocněním středních a malých arterií s postižením všech tří vrstev stěny cév vedoucích ke vzniku mnohočetných aneuryzmat, trombů a infarzací Etiopatogeneza: obvykle ? ? ? - úloha imunokomplexů, hypersenzitivní reakce, virové infekce Klinický obraz: variabilní – celkové příznaky + postižení vnitřních orgánů (kůže, klouby, ledviny, GIT, srdce…) Histologicky: postiženy tepny predilekčně v místě větvení fibrinoidní přestavba + infiltrace polymormonukleáry a eozinofily trombózy, aneuryzmata hojení vede k uzávěrům cév Glomerulonefritida – obvykle segmentová a proliferační

Pauci-imunitní nekrotizující GN Mikroskopická polyarteritida - postihuje cévy ledvin, event. Plic - obraz fokální segmentální nekrotizující GN s tvorbou srpků Wegenerova granulomatóza Nekrotizující zánět HCD (sinusitis) či DCD (ORL oblasti) + glomerulonefritida a systémová vaskulitida postihující kterýkoliv orgán ANCA – respirační vzplanutí fagocytujících buněk uvolnění kyslíkových radikálů degranulace poškození endoteliálních buněk (fokální proliferativní GNs tvorbou srpků)
 Etiopatogeneze: hyperglykémie ovlivňuje prostřednictvím glykace strukturu")
Diabetická nefropatie = diabetická interkapilární glomeruloskleróza (sy Kimmelstielův-Wilsonův) Etiopatogeneze: hyperglykémie ovlivňuje prostřednictvím glykace strukturu BM i mezangiální matrix průtoku plazmy se tlakem (hyperfiltraci) proliferace buněk ztluštění GMB s expanzí mezangia glomeruloskleróza Klinický obraz: stadium latentní (časné) – klinicky asymptomatické stadium incipientní DM nefropatie stadium manifestní DM nefropatie stadium chronické renální insuficince

Schematické znázornění průběhu diabetické nefropatie

Amyloidóza n Ledviny patří k orgánům nejčastěji postiženým oběma hlavními typy amyloidózy (tj. AL i AA amyloidózou) AL amyloidóza – komplikací monoklonálních gamapatií (myelomu, (primární) makroglobulinémie, některých lymfomů) AA amyloidóza – komplikace chronických zánětlivých onemocnění (RA, (sekundární) Bechtěrevovy choroby, TBC, Crohnovy nemoci apod. ) Klinický obraz: nefrotický syndrom, postupně se vyvíjí selhání ledvin
 Patogeneze: defekt syntézy")
Hereditární nefropatie Alportův syndrom - Hereditární nefritida s hluchotou (X vázaná) Patogeneze: defekt syntézy kolagenu ( -řetězců) GMB velmi tenká nebo mnohovrstevná GN fokální (difúzní) proliferacese segmentální sklerózou hematurie, proteinurie až závažné renální selhání (muži) Kongenitální nefrotický syndrom AR dědičnost Patogeneze: defekt syntézy složek bazálních membrán - masívní a neselektivní proteinurie Nefrotický syndrom of prvních týdnů života selhání ledvin -

Poruchy funkce tubulů Obecnými projevy porušené funkce tubulů jsou: n n n n n glykosurie při normální koncentraci glukózy v krvi aminoacidurie polyurie nadměrné ztráty Na+ (tzv. sůl ztrácející nefropatie) poruchy ve vylučování iontů H+ a a NH 3+ (tubulární acidóza) hypokalémie nebo hyperkalémie snížení koncentrační schopnosti ledvin přítomnost zvýšeného počtu válců v močovém sedimentu tubulární proteinurie ( 2 -mikroglobulin, lyzozym…)

Rozdělení poruch ledvinných tubulů n Vrozené poruchy tubulární metabolické defekty n polycystické onem. Ledvin Získané poruchy - poškození funkce při obstrukci v odtoku moče - ischemické poškození ledvinných tubulů - toxické poškození tubulů - poškození tubulů zánětem - poškození ledvinného intersticia při chron. Hyperkalcémii a hypokalémii - nefropatie způsobená analgetiky

Vrozené tubulární metabolické defekty n Fanconiho syndrom – důsledkem defektu Na+/K+-ATPasy - postiženy transportní funkce proximálního tubulu: * renální glykosurie (při normální glykémii) * tubulární proteinurie (především 2 -mikroglobulin) * aminoacidurie * hyperfosfaturie a hnypofasfatemie * hyperurikosurie * proximální renální tubulární acidóza ( resorpce hydrogenkarbonátů) * frakční exkrece Na+ > 1% - sekundární porucha kostí (rachitis, osteomalacie, poruchy růstu kostí) - tendence k hypovolémii (projevy ortostatické hypotenze) sekundární hyperaldosteronismus hypokalémie (sval. slabost)
 n n n RTA jsou nemoci, kdy ledviny nejsou schopny")
Renální tubulární acidózy (RTA) n n n RTA jsou nemoci, kdy ledviny nejsou schopny vylučovat do moči kyseliny, což vede k metabolické acidóze. Systémová acidóza s normálním aniontovým gapem (hyperchloremická), způsobená neschopností renálních tubulů udržovat acidobazickou rovnováhu, s normální GFR. Rozeznáváme typy 1 -4
 + K+(5) = Cl- (105)+HCO 3 -(25)")
Aniontový gap Ø Ø Ø Na+ (140) + K+(5) = Cl- (105)+HCO 3 -(25) + Gap (15) Gap se zvyšuje u metabolické acidózy, pokud dochází k přesunům iontů v extracelulární tekutině. Klinicky zajímavý, když se zvyšuje nad 30 Tkáňová hypoxie: laktát Diabetická ketoacidóza: 3 -hydroxybutyrát Renální selhání: sulfáty, fosfáty

 n Systémová acidóza s normálním aniontovým gapem (hyperchloremická), způsobená neschopností")
Renální tubulární acidózy (RTA) n Systémová acidóza s normálním aniontovým gapem (hyperchloremická), způsobená neschopností renálních tubulů udržovat acidobazickou rovnováhu, s normální GFR. n Rozeznáváme typy 1 -4 n Příčiny: - vrozené (genetické predispozice) - získané (sekundární): * imunologické * léky indukované * strukturální poškození tubulárních buněk
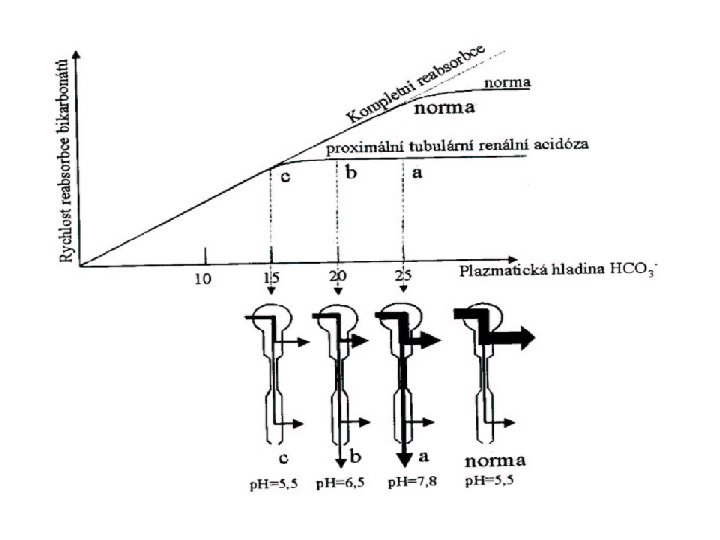
 - defektní je Na+/H+ kotransportér v prox. tubulu ( prox.")
Proximální RTA (typ II) - defektní je Na+/H+ kotransportér v prox. tubulu ( prox. sekrece H+) omezena resorpce bikarbonátů - většinou jen snížená hladina plazm. bikarbonátů (norm. kyselá moč) v krvi acidémie umožní reabsorbovat více Cl- hyperchlorémie - zvýšený přísun bikarbonátů (záporně nabité) do distálního tubulu sníží luminální potenciál – usnadní exkreci K+ hyperchloremická hypokalemická metabolická acidóza - v kombinaci s Fanconiho syndromem nebo izolovaně - zablokovat resorpci bikarbonátů můžeme i podáním diuretik blokujících karboanhydrázu

 - důsledkem defektu H+/K+-ATPasy v distálním tubulu a kortikálním sběracím")
Distální RTA (typ I) - důsledkem defektu H+/K+-ATPasy v distálním tubulu a kortikálním sběracím kanálku - je postižena acidifikace moči (v důsledku postižení sekrece H+ iontů do distálního nefronu) – moč se může okyselit jen na p. H = 5, 5 -6 - sníží se též reabsorpce bikarbonátů (norm. se vstřebává v dist. tubulu cca 10%) - za do moče ztracené bikarbonáty do krve přejdou chloridy - sníží-li se sekrece H+ iontů do tubulů, zvýší se tím exkrece K+ iontů hyperchloremická hypokalemická metbolická acidóza - další 2 důsledky distální RTA: - osteomalacie a hyperkalciurie – mobilizace solí Ca 2+ a Mg 2+ ze skeletu k pufrování kyselých aniontů - nefrokalcinóza a nefrolithiáza – acidózou podmíněné utlumení glykolyt. enzymů (např. fosfofruktokinázy) vede k potlačení renální tvorby a vylučování citrátu (pro vazbu Ca 2+) Ca 2+-fosfátové krystaly
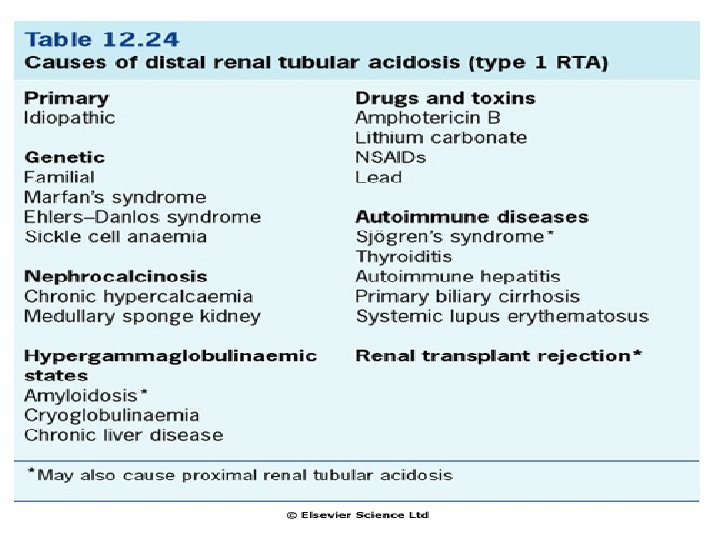

RTA – typ 4 n „Hyporeninemický hypoaldosteronismus“. - postižena resorpce Na+, exkrece K+ a sekrece H+ (aldosteron stimuluje sekreci H+ jednak stimulací H+ - ATPázového přenašeče a jednak nepřímo zvýšením resorpce sodíku v hlavních buňkách) hypochloremická acidóza s hyperkalémií n n - u pacientů s lehkou chronickou renální insuficiencí na podkladě tubulo-intersticiální nemoci ledvin (např. refluxní nefropatie) nebo diabetu. Plazmatický renin a aldosteron jsou nízké. Identický syndrom může být způsoben chronickým podáváním NSAIDs, které snižují sekreci reninu a aldosteronu. p. H moči nízké

Příčiny RTA – typu 4: n Deficit aldosteronu § § n Syndrom hyporeninemického hypoaldosteronismu Addisonova nemoc Heparin K+ nešetřící diuretika Dysfunkce sběracích kanálků při renální insuficienci § § Diabetická nefropatie Intersticiální nefritis Obstrukční uropatie Transplantace ledvin

 Proximální (II) Typ IV Útlum růstu ano ano K v séru")
Nález Distální (I) Proximální (II) Typ IV Útlum růstu ano ano K v séru N až p. H moči během acidózy >6 <6 <6 Exkrece K+ Exkrece Ca++ Exkrece citrátu Exkrece HCO 3 - při normálním <5 sérovém HCO 3(moč-krev) PCO 2 N až N (? ) N N > 15 < 15 N ? Glykosurie, aminoacidurie, hyperfosfatémie ne ano Ne Nefrokalcinóza ano ne Ne Křivice ano ne ne Doporučení pro K+ ne zvýšit ne
 n Spočívá v omezení dostupnosti amoniaku v distálním")
RTA při snížené GF (typ III) n Spočívá v omezení dostupnosti amoniaku v distálním nefronu n (chybí akceptor secernovaných H+ iontů, čímž se snižuje gradient H+, což umožňuje zvýšit účinnost H+ pumpy) Exkrece H+ iontů (a ekvimolární přísun bikarbonátů z ledvin do krve) se proto snižuje, bikarbonáty jsou nahrazeny zvýšenou resorpcí chloridů hyperchloremická acidóza (normokalemická s norm. deficitem aniontů) n Hlavní příčina: nedostatečná funkce protiproudového systému Henleho kličky - postižení intersticia ledvin zánětem - pokles GFR do 20 ml/min (při výraznějším poklesu GFR se hromadí i anionty silných kyselin uremická normochloremická acidóza se zvýšeným deficitem aniontů a hyperkalémií)

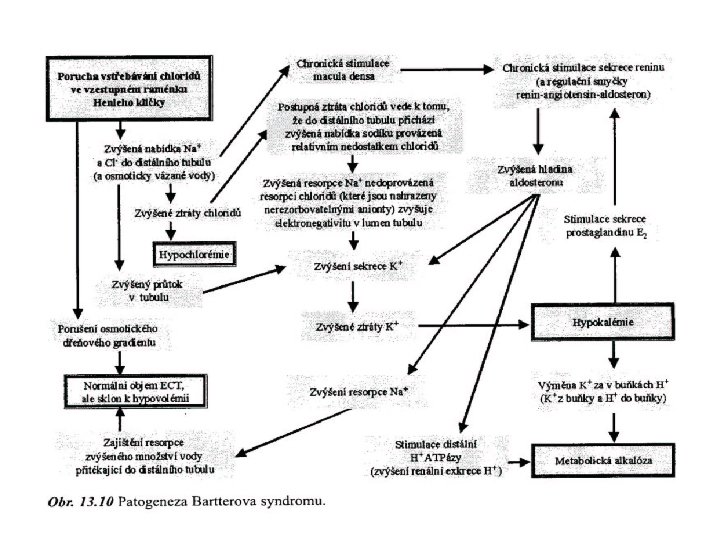
Bartterův syndrom n n n Příčinou je vrozený defekt Na+/K+/2 Cl- symportu v epiteliích vzestupné části Henleovy kličky Snížená resorpce Na+ vede ke zvýšenému Na+ v distálních segmentech, což vede k stimulaci resorpce Na+ v distálním tubulu s následnou zvýšenou sekrecí K+ vylučování PGE 2 stimulaci reninu zvýšení produkce ANG II a aldosteronu (zesiluje sekreci K+) Projevy: sekundární hyperaldosteronismus se značnou hypokalémií a metabolickou alkalózou svalová slabost Pseudo-Barterrův syndrom - při chronickém užívání diuretik „nešetřících K+“ – např. furosemidu

Liddlův syndrom n n Jde o defekt apikálních Na+ kanálů v distálním tubulu, které jsou norm. aktivovány aldosteronem – při této poruše jsou aktivovány trvale (označován jako psudohyperaldosteronizmus) dochází k zesílené resorpci Na+ a sekreci K+ a H+ důsledkem je hypertenze, hypokalémie a alkalóza Na+ kanál se dá terapeuticky selektivně blokovat diuretikem zv. amilorid
 n n Označuje se i jako vitamin-D rezistentní rachitis Příčina:")
Vrozená hypofosfatémie (fosfátový diabetes) n n Označuje se i jako vitamin-D rezistentní rachitis Příčina: defekt 2 Na+HPO 42 - kotransportéru v proximálním tubulu (a v jejunu) - způsobuje osteomalácii - jako izolovaná porucha - v rámci Fanconiho syndromu
 nebo")
Renální diabetes insipidus - defekt genu pro V 2 receptor ( X chromozom) nebo mutace genu pro vodní kanál akvaporin 2 v epiteliích sběr. kanálků snížená schopnost koncentrovat moč i při dostatku ADH polyurie s nízkou osmolalitou moče dehydratace (s hypernatrémií)
 n n Porušena resorpce glukózy (objevuje se v moči")
Renální glukosurie (renální diabetes mellitus) n n Porušena resorpce glukózy (objevuje se v moči už při malém zvýšení koncentrace) Rozlišujeme 2 typy: - typ A: snížená maximální transportní rychlost pro glukózu - typ B: snížená afinita transportéru pro substrát Příznaky: - glukosurie - osmotická diuréza typ B se může vyskytovat součastně s transportní poruchou ve střevě

Poruchy transportu aminokyselin n n většinou jsou postiženy kotransportéry na apikálních membránách často nejen v ledvinách, ale i v játrech a ve střevě aminoacidurie n koncentrace AK ve stolici poruchy jater Koncentrace AK v plazmě – normální (postižena jen renální resorpce) - snížená Hartnupova nemoc - defekt transportérů pro neutrální AK (mimo glycin a iminokys. ) - důsledky: projevy nedostatku AK, poškození CNS produkty odbourávání (např. indikán), snížená syntéza nikotinamidu – kožní pelagroidní změny

Klasická cystinurie - - Defekt resorpce lysinu, argininu a ornithinu - hromadí se v lumen tubulů a tlumí resorpci cystinu Projevy: vznik konkremetů v ledvinách (AK s nejnižší rozpustností) Malabsorpce tryptofanu (sy modrých plenek) - v důsledku oxidace na indikán Malabsorpce methioninu a fenylalaninu (syndrom sušárny chmele) - typický pach moči viz výše
 onemocnění s tvorbou")
Polycystické onemocnění ledvin n jde o AD (vzácněji AR - juvenilní) onemocnění s tvorbou mnohočetných cyst s epitelovou výstelkou v ledvinách (vzácněji i játrech) Mutace genů PKD 1 a PKD 2 – produkty jsou polycystin 1 a 2 (funkce není dostatečně známa – snad plní funkci kanálů pro kationty (Ca 2+) v membráně ER) Projevy: - zvětšení ledvin - hematurie - renální arteriální hypertenze CHSL se sníženou koncentrační schopností ledvin a GFR

Získané tubulární metabolické defekty n n Při poškození tubulů jde většinou i o postižení intersticia ledvin tubulointersticiální nefropatie (tubulointersticiální nefritida) Projevy: * počáteční hypertrofie tubulů, přítomnost zánětlivých buněk v intersticiu, proliferací fibroblastů a hromaděním kolagenu * změny vyústí v atrofii tubulů a fibrotizaci intersticia CHSL Významná role v patogenezi se přisuzuje angiotenzinu II farmaka inhibující tvorbu ANG II zpomalují průběh onemocnění

Předpokládané účinky angiotenzinu II v patogenezi tubulointersticiální nefropatie

Poškození funkce ledvinových tubulů při obstrukci v odtoku moči n tlaku v ledvinné pánvičce a ledvinných tubulech vede k dilataci interference s aktivním transportem moči ( tlak se propaguje do Bowmanova pouzdra GFR RBF atrofie nefronů Klinickou manifestací: anurie a ASL při úplné oboustranné obstrukci CHSL s polyurií, azotémíí, hyperkalémií u částečné Po uvolnění obstrukce může dojít k tzv. postobstrukční diuréze (výrazem obnovení nebo zvýšení GFR při současném postižení tubulů)
")
Ischemické poškození ledvinových tubulů n Jde o stav označovaný jako akutní tubulární nekróza (ATN)
")
Toxické poškození ledvinových tubulů n Celá řada nefrotoxických látek – zánětlivá reakce (intersticiální nefritis) - látky s původem v organizmu: hemoglobin, myoglobin, kys. Močová - kovy: rtuť, arzen, olovo, kadmium… - tuková rozpouštědla: toulen, etylenglykol… - cytostatika: cisplatina, metotrexát… - ATB: amfotericin B, aminoglykosidy, TTC, sulfonamidy… - diuretika: furosemid, tiazidy - jiné: amfetaminy, ACEI, cyklosporin… Poškození ledvin může být – akutní (ATN) ASL - chronické CHSL

Poškození ledvinových tubulů zánětem n n Chronický zánět - zdroj kyslík. radikálů, proteáz, komplementu, cytotoxických lymfo, cytokinů…. (poškození) Může jít o: a) abakteriální zánět – může být součástí toxického poškození b) alergický zánět c) bakteriální zánět - šířící se ascendentně (pyelonefritida) ! - descendentní cesta (hematogenní) predispozicí : poruchy odtoku moči DM

Poškození intersticia ledvin při chronické hyperkalcémii a hypokalémii n Nefropatie při chronické hyperkalcémii: - z důvodu hyperparatyreózy, sarkoidózy, nádor. onemocnění, intoxikace vit. D… - způsobena kalcifikacemi v oblasti sběracích kanálků, distálního nefronu a Henleovy kličky dilatace a atrofie tubulů a postižení intersticia - je snížena koncentrační schopnost ledvin polyurie, nykturie (CHSL) n Nefropatie při chronické hypokalémii: - může způsobit snížení koncentrační schopnosti ledvin (narušení mechanizmu tvorby osmotického gradientu v intersticiu dřeně ledviny)

Nefropatie způsobená analgetiky n Chronické užívání kombinace analgetik obsahujících směs kyseliny acytylosalicylové paracetamolu kofeinu může poškodit kapiláry ve dřeni ledvin a vést k nekróze renální papily * hematurie * ledvinná kolika - závažná je intersticiální fibróza a atrofie kůry ledviny
- Slides: 74