MYELOPROLIFERATIVE NEOPLASMS MPN Hematologic Malignancies Classification Lymphoid Myeloid
 경희의대 종양혈액내과 조 경 삼")

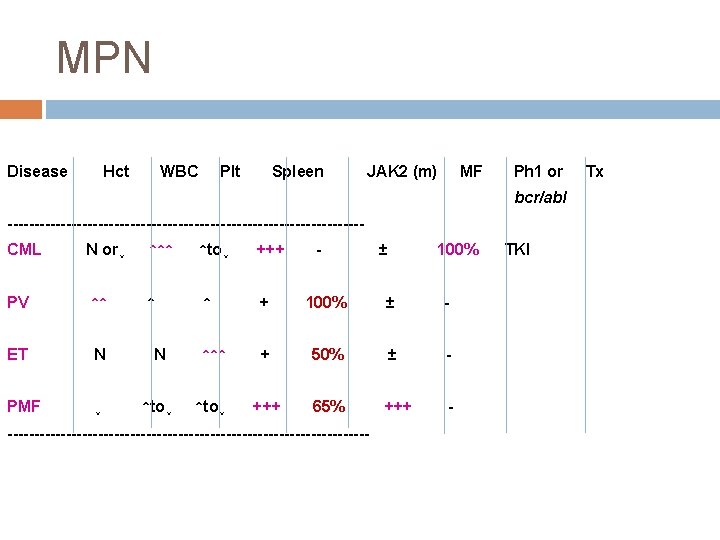
 Classification Chronic myelogenous leukemia, BCR-ABL 1 positive (CML) Polycythemia vera (PV)")
 특성 Chronic myeloid disorder Molecular changes � � Overproduction of blood cells without")

 만성골수성백혈병")
 Definition: Clonal")
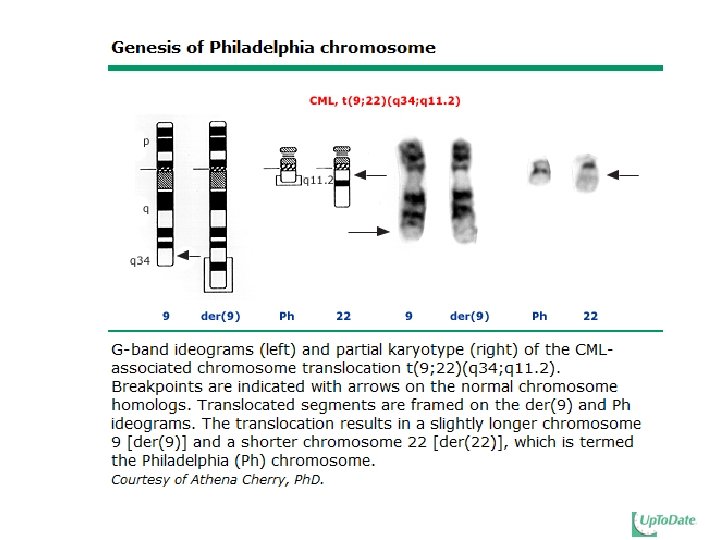
(q 34; q 11) BCR/ABL 1 fusion gene BCR-ABLl fusion")

 Leukocytosis: left")
 Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Atypical CML")


 : � Dasatinib, Nilotinib,")




 Ø Ø Ø t(9; 22)(q 34; q 11), BCR-ABL 1 Chronic, accelerated,")
 진성적혈구증가증")


, Weakness (47%), Dizziness (43%) excessive sweating")
 Splenomegaly (70%) Facial plethora (ruddy cyanosis) (67%) Hepatomegaly (40 percent) Injection")

 Criteria PVSG (1967) WHO (2008) Major M 1: increased red")

: marrow fibrosis, marked splenomegaly, anemia")
 진성혈소판증가증")


 BM: large numbers of hyperploid megakaryocytes, progressing")

 Iron-deficiency anemia Hyposplenism Postsplenectomy Malignancy Collagen vascular diseases Inflammatory bowel disease")
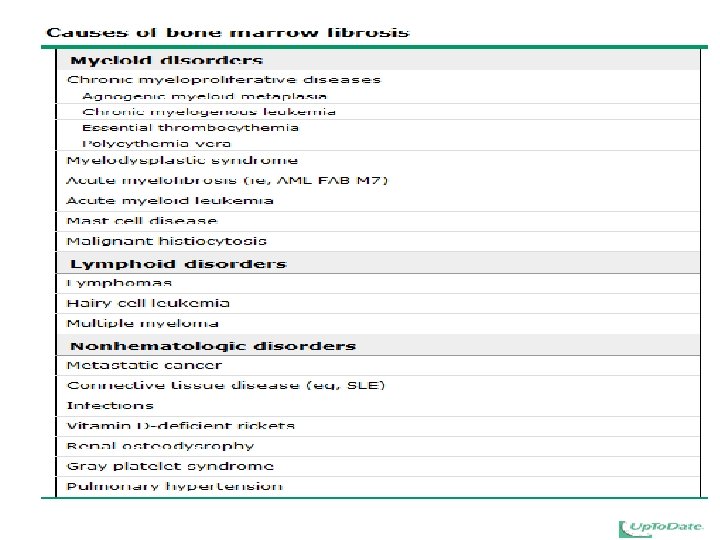
 Primary myelofibrosis ET CML Idiopathic sideroblastic anemia Myelodysplasia (5 q- syndrome)")


 일차골수섬유증")
 Idiopathic myelofibrosis (IMF, AMM/MF) Clonal disorder of the hematopoietic stem cell characterized")



 RBC: teardrop cell (dacrocyte), fragmented cells and nucleated red cell, initially mild")
 Bone sclerosis (50%) Spleen: massive enlargement Extramedullary hematopoiesis: spleen, kidney, lung, adrenal")



 Hydroxyurea JAK 2 inhibitor (Ruxolitinib) Others � Alkylating")


- Slides: 58
MYELOPROLIFERATIVE NEOPLASMS (MPN) 경희의대 종양혈액내과 조 경 삼
Hematologic Malignancies Classification Lymphoid, Myeloid Acute, chronic Chronic myeloid disorders � Dysplastic : MDS � Proliferative : MPN � MDS/MPL : CMML Juvenile CMML Atypical CML Unclassifiable MDS/MPL
Myeloproliferative neoplasms (MPN) Classification Chronic myelogenous leukemia, BCR-ABL 1 positive (CML) Polycythemia vera (PV) Primary myelofibrosis (PMF) Essential thrombocythemia (ET) Chronic neurophilic leukemia (CNL) Chronic eosinophilic leukemia, NOS (CEL) Mastocytosis � Cutaneous mastocytosis � Systemic mastocytosis � Mast cell leukemia � Mast cell sarcoma � Extracutaneous matocytoma Myeloproliferative neoplasm, unclassifiable (MPN, U)
MPN(CMPD) 특성 Chronic myeloid disorder Molecular changes � � Overproduction of blood cells without significant dysplasia (effective production) Predilection to extramedullary hematopoiesis Myelofibrosis : TGF-beta, marrow failure Clonal evolution � CML : t(9; 22), bcr-abl PV, ET, PMF : JAK 2, CALR, MPL, TET 2, etc. MDS/AML Thrombosis and bleeding
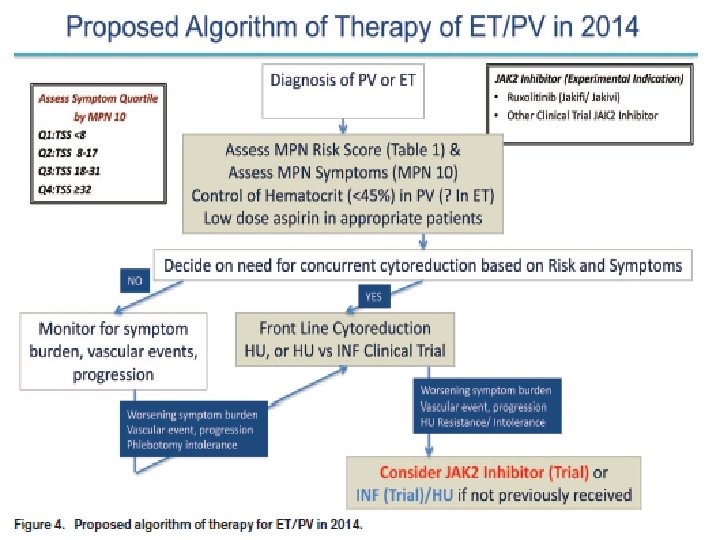
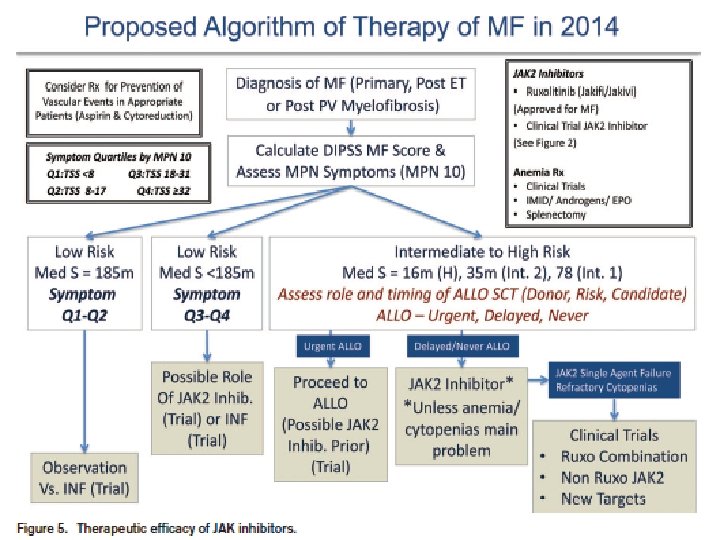
MPN 치료 Cure Survival prolongation Symptom control � MPN-SAF Prognoatic factors Risk benefit Cost effectiveness Observation Medical Treatment Cytoreductive � Target � Allogeneic stem cell transplantation
CHRONIC MYELOGENOUS LEUKEMIA (CML) 만성골수성백혈병
Chronic myelogenous leukemia (CML, also known as chronic myelocytic, chronic myeloid leukemia) Definition: Clonal myeloproliferative disorder of a pluripotent hematopoietic stem cell with a specific cytogenetic abnormalities, Philadelphia Chromosome (Ph) t(9; 22)(q 34; q 11) BCR-ABL 1 fusion gene results in the formation of a unique gene product, the BCR-ABL 1 fusion protein (tyrosine kinase). Chronic, accelerated, and blastic phases At any age, but peak in the age group of 50 - 60
Pathophysiology Philadelphia chromosome: t(9; 22)(q 34; q 11) BCR/ABL 1 fusion gene BCR-ABLl fusion protein (p 210 BCR-ABL) normal abl gene: p 145, tyrosine kinase Result of BCR-ABL 1 activity Uncontrolled proliferation of transformed cells Discordant maturation Escape apoptosis Altered interaction with cellular matrix
Symptoms and signs Asymptomatic : leucocytosis Splenomegaly, hepatomegaly, rare lymphadenopathy Hypermetabolism: weight loss, fatigue, elevated uric acid Bone pain, arthralgia, pain from splenic infarction Thrombohemorrhagic complication fever,
Lab findings Ph : chromosome analysis or FISH BCR/ABL : RT-PCR, quantitation(Q-PCR) Leukocytosis: left shift, eosinophilia, basophilia Thrombocytosis: normal morphology and function Hyperuricemia Gaucher cells or see-blue histiocytes in BM Increased vitamin B 12 and binding capacity Reduced LAP score
D Dx Leukemoid reactions (infection, neoplasms) Juvenile myelomonocytic leukemia Chronic myelomonocytic leukemia Atypical CML Chronic eosinophilic leukemia Chronic neutrophilic leukemia Other MPN Other Philadelphia chromosome-positive malignancies ALL � AML �
Natural course of CML Chronic phase � uncontrolled production of mature and maturing granulocytes, predominantly neutrophils, but also basophils and eosinophils Intermediate, accelerated phase � neutrophil differentiation becomes progressively impaired and leukocyte counts are more difficult to control with treatment Blastic (acute transforming) phase �a condition resembling acute leukemia in which myeloid or lymphoid blasts proliferate in an uncontrolled manner
Prognostic scoring system Sokal prognostic score � spleen size, percent blasts, age, and platelet count >700, 000/μL Hasford or Euro score � adds eosinophilia and basophilia EUTOS score � after imatinib (glivec) � the percentage of basophils and spleen size
Treatment TKI : tyrosine kinase inhibitor � Imatinib mesylate (Glivec®) : � Dasatinib, Nilotinib, Bosutinib, � Ponatinib (T 315 I mutation) Radotinib � Response : MMR(major molecular remission) � Duration of treatment : ? � Resistance, progression or relapse � Toxicity management Allogeneic stem cell transplantation (NCCN guidline 참고)
Resistance to TKIs Bcr-Abl mutation � � The T 315 I mutation : HSCT, ponatinib, omacetaxine, or clinical trials The Y 253 H, E 255 K/V, or F 359 V/C/I mutations : dasatinib The F 317 L/V/I/C, V 299 L, or T 315 A mutations : nilotinib The E 254 K/V, F 317 L/V/I/C, F 359 V/C/I, T 315 A, or Y 253 H : bosutinib Bcr-Abl amplification Enhanced expression of multidrug exporter proteins Alternative signaling pathway Clonal cytogenetic evolution
Second line treatment Palliative therapy � IFN-α: sometimes cytogenetic remission � Other cytoreductive chemotherapy: Hydroxyurea Busulfan Cytosine arabinoside Omacetaxine (HHM : homoharringtonine) � Splenectomy or splenic irradiation � Blastic phase: Induction chemotherapy for acute leukemia, SCT � RT for extramedullary tumor
SCT and DLI Allogeneic SCT � HLA-matched sibling � Mismatched relatives � Matched unrelated � Cord blood Autologous SCT : not curative DLI NMAST (mini-transplantation)
Summary (CML) Ø Ø Ø t(9; 22)(q 34; q 11), BCR-ABL 1 Chronic, accelerated, and blastic phases TKI : tyrosine kinase inhibitor Ø Ø Ø Imatinib mesylate (Glivec®) : Dasatinib, Nilotinib, Bosutinib, Radotinib Ponatinib (T 315 I mutation) Duration of treatment : ? Allogeneic stem cell transplantation : Cytoreductive or Symptomatic Tx
POLYCYTHEMIA VERA (PV) 진성적혈구증가증
Polycythemia vera Definition: Myeloproliferative disorder resulting from clonal expasion of a transformed hematopoietic stem cell associated with prominent over-production of erythrocytes and to lesser extent expansion of granulocytic and megakaryocytic elements Gradual in onset and runs a chronic but usually slowly progressive Generally benign in late middle life, slightly more common in males Expected survival less than 2 years if not treated
Etiology Unknown 20 q-, trisomy 8, 9 : 30% JAK 2 mutation (V 617 F; val 617 phe) : nonreceptor tyrosine kinase : gain of function : 30% homozygosity JAK 2 12, MPL, CALR, TET 2, etc. Hypersensitivity to IGF-I, IL-3, GM-CSF : PV, ET Up-regulation of bcl-XL : antiapoptotic PVR-1 m. RNA : increased
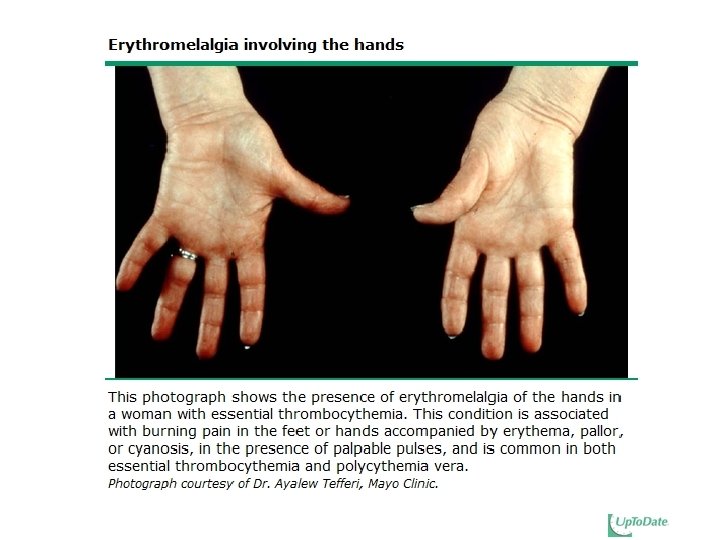
Clinical features Nonspecific complaints : � Headache (48%), Weakness (47%), Dizziness (43%) excessive sweating (33%) Acute gouty arthritis --- 5 to 20% Pruritus --- 31% Erythromelalgia Thrombosis � CVA, MI, superficial thrombophlebitis, DVT, PE � Budd-Chiari syndrome, and portal, splenic, or mesenteric vein thrombosis � Transient visual disturbances (eg, amaurosis fugax, scintillating scotomata, ophthalmic migraine) Gastrointestinal symptoms � Peptic ulcer, epigastric distress, gastroduodenal erosions
Clinical features (Signs) Splenomegaly (70%) Facial plethora (ruddy cyanosis) (67%) Hepatomegaly (40 percent) Injection of the conjunctival small vessels and/or engorgement of the veins of the optic fundus Excoriation of the skin, which might be extensive, suggesting the presence of severe pruritus Sequellae of arterial or venous thrombotic event (eg, stroke, DVT, superficial thrombophlebitis) Gouty arthritis and tophi Hypertension(1/3): systolic
Lab findings Elevated Hb, normocytic normochromic, polychromasia, nucleated RBC Leukocytosis, basophilia, increased LAP, serum vitamin B 12, and binding capacity Thrombocytosis, defective platelet function BM: erythroid hyperplasia or panhyperplasia, iron may be absent, mild fibrosis Cytogenetic abnormalities (up to 30%): trisomy 1, 8, or 9 and 20 q. Low Epo level : normal 4 -26 m. U/m. L JAK 2 mutation � � Exon 14 : JAK 2 V 617 F Exon 12 (97%) (3%)
PV 진단 (WHO 2008) Criteria PVSG (1967) WHO (2008) Major M 1: increased red cell mass (>36 ml/kg in men, >32 ml/kg in women) M 2: Normal oxygen saturation (>92%) M 3: Splenomegaly; A 1: Elevated red cell mass >25% or Hb > 18. 5 g/d. L (men), 16. 5 g/d. L (women) or 17 g/d. L (men), 15 g/d. L (women), if increase of ≥ 2 g/d. L A 2: Presence of JAK 2 V 617 F mutation or similar mutation such as JAK 2 exon 12 mutation Minor thrombocytosis (>400, 000/ L) and leukocytosis (12, 000/ L) LAP Vit B 12, binding capacity B 1: BM hypercullarity B 2: Low serum EPO B 3: EEC(endogenous erythroid colony) formation in vitro Diagnosis M 1+ M 2 + M 3 M 1+ M 2 + 2 minor A 1 + A 2 + any B or A 1 +any two B
Therapy No therapy: 6 – 18 months Phlebotomy: 10 -12 years Myelosuppression: � Phlebotomy should be used initially � Chemotherapy: Hydroxyurea JAK 2 inhibitors (Ruxoritinib) : failed hydroxyurea IFN-alpha Anagrelide for thrombocytosis alkylating agent: busulfan, melphalan, chrlorabucil Pipobroman � RT: 32 P : 6 -24 months remission, 2 nd Leukemia Cyproheptadine, hydroxyzine, cimetidine for pruritus Allopurinol for hyperuricemia
Course and prognosis Vascular complication: majority Post-PV MF(15 -20%): marrow fibrosis, marked splenomegaly, anemia Secondary hematologic neoplasm: acute leukemia(1 -2%), NHL, MM Increased secondary leukemia with 32 P and alkylating agents
ESSENTIAL THROMBOCYTHEMIA (ET) 진성혈소판증가증
Essential thrombocythemia Markedly elevated platelet production in the absence of recognizable stimuli Blood platelet counts above 600, 000 may reach levels of 3 to 4 million/µL Clinical involvement of hematopoietic lineages other than megakaryocytic line is unusual Clonal disorder of pluripotent hematopoietic stem cell Autonomous in vitro megakaryocytic colony formation
Clinical features 2/3: asymptomatic at diagnosis Vasomotor symptoms Headache � Lightheadedness � Syncope � Atypical chest pain � Acral paresthesia � Livedo reticularis � Erythromelalgia � Transient visual disturbances � Thrombosis Bleeding
Lab findings Thrombocytosis, giant hypogranular, abnormal function(epinephrine) BM: large numbers of hyperploid megakaryocytes, progressing fibrosis JAK 2 V 617 F mutation : 50 % Cytogenetic abnormalities: uncommon
Diagnostic criteria Sustained platelet count >450, 000/u. L. Bone marrow biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes; no significant increase or left-shift of neutrophil granulopoiesis or erythropoiesis. Not meeting WHO criteria for PV, PMF, CML, MDS of other myeloid neoplasm. Demonstration of JAK 2 V 617 F mutation or other clonal marker, or in the absence of a clonal marker, no evidence for reactive thrombocytosis.
Causes of thrombocytosis(I) Iron-deficiency anemia Hyposplenism Postsplenectomy Malignancy Collagen vascular diseases Inflammatory bowel disease Infection Hemolysis Hemorrhage PV
Causes of thrombocytosis(II) Primary myelofibrosis ET CML Idiopathic sideroblastic anemia Myelodysplasia (5 q- syndrome) Postsurgery Rebound: cessation of ethanol intake, correction of vitamin B 12 or folate deficiency
Therapy Myelosuppression: hydroxyurea, alkylating agents, 32 P IFN-alpha Anagrelide Emergency plateletpheresis Platelet transfusion Aspirin and dipyridamole: protect thrombosis but serious bleeding may occur
Course and prognosis Survival will be at least as good as for those of PV Transform to a more aggressive or frankly leukemic phase (5%) Progress to MF (25%)
PRIMARY MYELOFIBROSIS (PMF) 일차골수섬유증
Primary myelofibrosis(PMF) Idiopathic myelofibrosis (IMF, AMM/MF) Clonal disorder of the hematopoietic stem cell characterized by BM fibrosis and extramedullary hematopoiesis In the late middle life, gradual onset, and progressive
Etiology 9 p, 20 q–, 13 q–, trisomy 8 or 9, or partial trisomy 1 q are common Fibrosis in this disorder is associated with overproduction of TGFβ and tissue inhibitors of metalloproteinases, osteosclerosis is associated with overproduction of osteoprotegerin, an osteoclast inhibitor Marrow angiogenesis occurs due to increased production of VEGF Fibroblasts in chronic IMF are polyclonal
Clinical features Vague constitutional symptoms associated with anemia, fatigue, weight loss, night sweat Splenomegaly, hepatomegaly, lymphadenopathy Thrombocytopenia: petechia, bleeding Extramedullary hematopoiesis : jaundice, ascites, bone pain, portal, pulmonary, intracranial hypertension, intestinal or ureteral obstruction Splenic infarction : fever, pleuritic chest pain Hyperuricemia, secondary gout
Clinical features Generalized Sx : fatigue, wt loss, mild fever, night sweat Thrombotic events : incidence is same as ET Splenomegaly : sometimes massive Hepatomegaly : sometimes potal hypertension Extramedulary hematopoiesis : in any organ � � � � In or surrounding the vertebral column (especially thoracic) Lymph nodes Retroperitoneum Lungs or pleura Genitourinary system Skin Other sites (thalamus, right atrium, mouth, muscle, bowel) Bone and Joint involvement : osteosclerosis, periosteitis, pain and tenderness
Lab findings(I) RBC: teardrop cell (dacrocyte), fragmented cells and nucleated red cell, initially mild anemia but progressing(ineffective erythropoiesis and decreased red cell survival) WBC: leukopenia (25%) to leukocytosis, shifting to left and circulating blasts, basophilia Increased circulating CD 34+ cells Platelet: normal or elevated but eventually thrombocytopenia, giant form platelets or megakaryocytes, abnormal platelet function
Lab findings(II) Bone sclerosis (50%) Spleen: massive enlargement Extramedullary hematopoiesis: spleen, kidney, lung, adrenal gland, LN BM: dry tap, progressive fibrosis and osteosclerosis Cytogenetic abnormalities: 9 p, 20 q-, 13 q-, trisomy 8 or 9, partial triosomy 1 q
Diagnosis Bone marrow biopsy: essential May be difficult to distinguish from other MPN Rule out other causes of myelofibrosis Acute myelofibrosis: M 7 Myelodysplasia with myelofibrosis
Diagnostic criteria Major criteria � Presence of megakaryocyte proliferation and atypia, usually accompanied by either reticulin and/or collagen fibrosis, or, in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased BM cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (ie, prefibrotic dellularphase disease. � Not meeting WHO criteria for PV, CML, MDS of other myeloid neoplasm. � Demonstration of JAK 2 V 617 F mutation or other clonal marker (eg, MPL W 515 K/L), or in the absence of a clonal marker, no evidence of BM fibrosis due to underlying inflammatory of other neoplastic diseases. Minor criteria � Leukoerythroblastosis � Increase in serum LDH � Anemia � Palpable splenomegaly All 3 major criteria and 2 minor criteria
Modified IPSS Dynamic IPSS DIPSS Plus Age> 65 -- ------ 1 WBC count > 25, 000/ u. L – --1 Hb < 10 g/d. L – ------2 Circulating blast cell ≥ 1% - 1 Presence of constitutional symptom – -------1 0: low 1 -2 : Intermaeiate-1 3 -4 : Intermediate-2 5 -6 : high DIPSS low –-------- 0 DIPSS Int-1 –------- 1 DIPSS Int -2 –------ -- 2 DIPSS high –------- - 3 Unfavorable kqryotype –--- 1 Platelet < 100, 000/u. L –---- 1 Transfusion need –----- 1 0: 1 -2 : 3 -4 : 5 -6 : low Intermaeiate-1 Intermediate-2 high
Therapy BMT (allogeneic stem cell transplantation) Hydroxyurea JAK 2 inhibitor (Ruxolitinib) Others � Alkylating agents � Thalidomide plus prednisone � Lenalidomide (symptomatic patient with 5 q-) � Etanercept Radiation : spleen, other EMH areas Splenectomy Androgens, danazol (anemia) Blood transfusions with or without erythropoietin (anemia)
Course and prognosis Median survival: 4 -5 years, 25% 15 years Major causes of death: infection, CHF, renal failure, portal hypertension, hemorrhage Acute leukemia: 5 -10% IPSS prognostic factor � � � Presence of constitutional symptoms (ie, weight loss >10 percent, night sweats, or fever) Age >65 years Hemoglobin <10 g/d. L Leukocyte count >25, 000/micro. L Circulating blast cells ≥ 1 percent
A genetic and histology‑based diagnostic algorithm for CML, PV, ET and PMF