Musculoskeletal diseases Lecture 2 Disorders of skeletal muscle









")


 Muscular dystrophies: Dystrophinopathies: They are primary M. degeneration.")







.")

. 4. Due")


- Slides: 29
Musculoskeletal diseases Lecture 2 Disorders of skeletal muscle
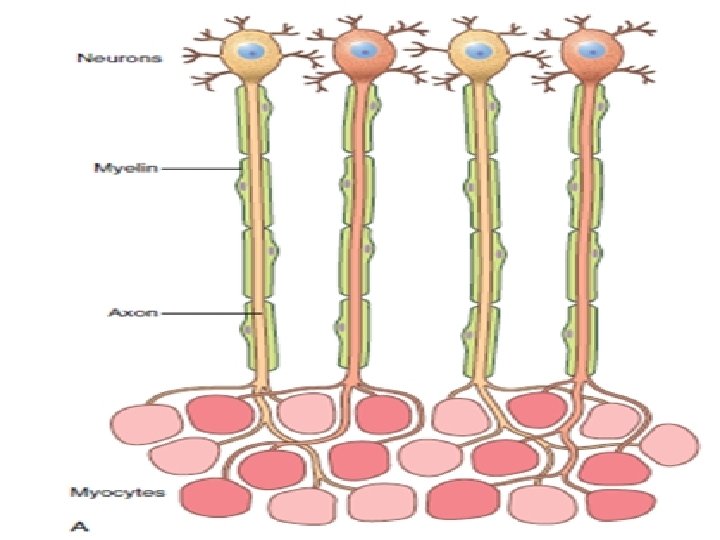
The motor unit consists of : 1 -Motor neuron in the CNS. 2 -Peripheral axon. 3 -Neuromuscular junction. 4 -Skeletal muscle fibers: Two type: 1 - Slow twitch type I. (Faint) 2 - Fast twitch type II fibers. (Dark) Distributed in checkerboard pattern.
Normal skeletal muscle: Uniform polygonal myofibers, peripherally placed nuclei, tightly packed together into fascicles separated by scant connective tissue (Perimysium). Perimysial interfascicular septum containing blood vessel) (arrow)
II I ATPase stain
Classification of muscle diseases: Myopathies 1 - Primary myopathies. 2 - Secondary: Neuropathic changes caused by disorders in muscle innervation. Both Muscle fiber atrophy. 1. Primary myopathies: Morphology: 1 -Segmental necrosis & regeneration of muscle fibers. 2 -Inflammatory cells infiltration. 3 -Disruption of muscle by fibrosis & fat replacement. (Features of disease chronicity)
B 1 B 2 B 3 B 4 Skeletal myopathy: v. Segmental necrosis & regeneration of individual myofibers. v. Necrotic cells (B 1 -B 3): Infiltrated by infl. cells. v. Regenerative myofibers (B 4, arrow): Cytoplasmic Basophilia & atrophy.
2. Neuropathic Atrophy: Type of myofibers is not an inherent feature but determined by: Innervating motor neuron. Injury & regeneration of peripheral nerves Alters muscle innervation Changes in distribution of type I & type II myofibers. In skeletal muscle disorders caused by abnormal innervation: 1. No degeneration & regeneration of individual fibers. 2. No inflammatory cells infiltrates.
Neuropathic Atrophy: Characterized by: A- Fiber type grouping: Changes in muscle innervation presence of larger groups of fibers of same type. Replacement of normal checkerboard distribution by groups of type I or type II fiber B- Grouped atrophy: Changes & separation of innervated fibers in fewer but larger motor units results in groups of atrophic fibers.
Features of neurogenic myopathy: Fiber-type grouping: Patchy areas of myofibers sharing the same fiber type. ATPase reaction: Method of distinguishing between fiber type: Type I stain more lightly than type II fibers. Loss of “checkerboard” pattern
Clusters of atrophic myofibers (Grouped atrophy)
Another causes of M. atrophy: 1. Prolonged disuse of muscles: Cause focal or generalized muscle atrophy. E. x. Prolonged bed rest. Casting of broken bone 2. Glucocorticoid exposure: Exogenous or endogenous (e. g. , in Cushing syndrome), cause muscle atrophy.
Classification of Skeletal Muscles Diseases Inherited Disorders. Acquired Disorders.
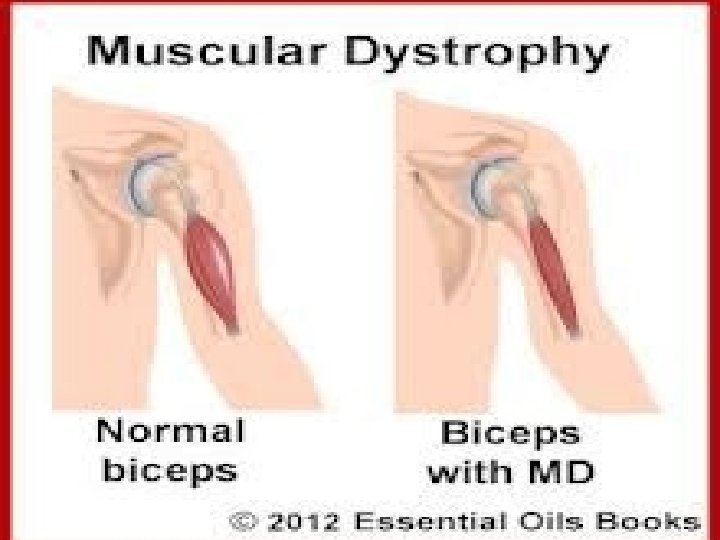
Inherited Disorders of Skeletal Muscle: (Genetic) Muscular dystrophies: Dystrophinopathies: They are primary M. degeneration. Inherited diseases result in progressive muscle injury in patients appears normal at birth. Most common form of muscular dystrophy: Duchenne muscular dystrophy (DMD). Becker muscular dystrophy (BMD).
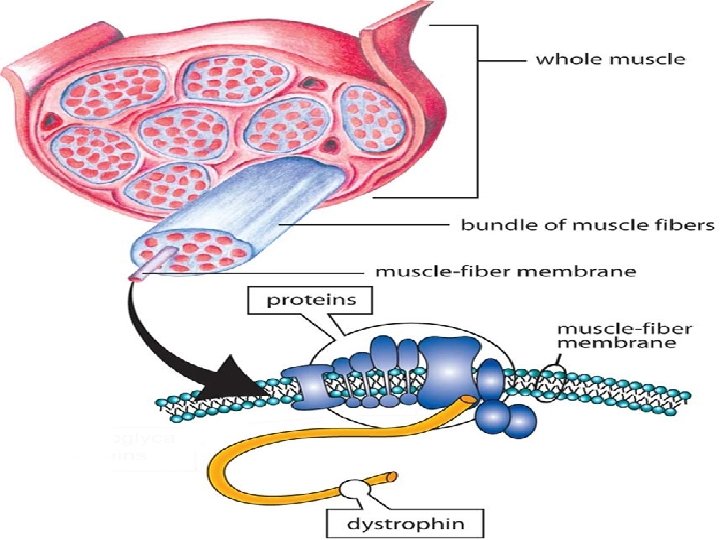
Pathogenesis: Both DMD & BMD are caused by: Mutations in dystrophin gene located on short arm of X chromosome (Xp 21).
Dystrophin: Very large protein found in skeletal & cardiac muscle, brain, & peripheral nerves. It is part of dystrophin-glycoprotein complex that stabilizes muscle cell during contraction. Function of types I & II fibers depends on this protein complexes. So absence of dystrophin will cause impaired contractility.
Defects in this complex: Transient membrane tears of muscle cells during contractio myofiber degeneration, including cardiac muscle cardiomyopathy. In DMD: Complete absence of dystrophin. In BMD: Defective form of dystrophin. Thus, severity of disease correlates with degree of dystrophin deficiency.
Duchenne muscular dystrophy: Clinically evident by age of 5 years. Patients are wheelchair-bound at teenage. Dead by early adulthood.
Morphology Histology: Skeletal muscles changes in DMD & BMD are similar but milder in BMD. Myofiber necrosis & regeneration. Progressive replacement of muscle T. by fibrosis & fat. Muscles shows variation in myofiber size & abnormal internally placed nuclei.
Early stage disease: Fascicular muscle architecture is maintained. Myofibers show variation in size. Cluster of basophilic regenerating myofibers (arrow). Slight endomysial fibrosis. (Focal pink-staining C. T. between myofibers).
Disease progression: Extensive variation in myofiber size. Fatty replacement. Endomysial fibrosis.
Immunohistochemical staining shows complete absence of membrane-associated dystrophin, seen as brown stain (arrow).
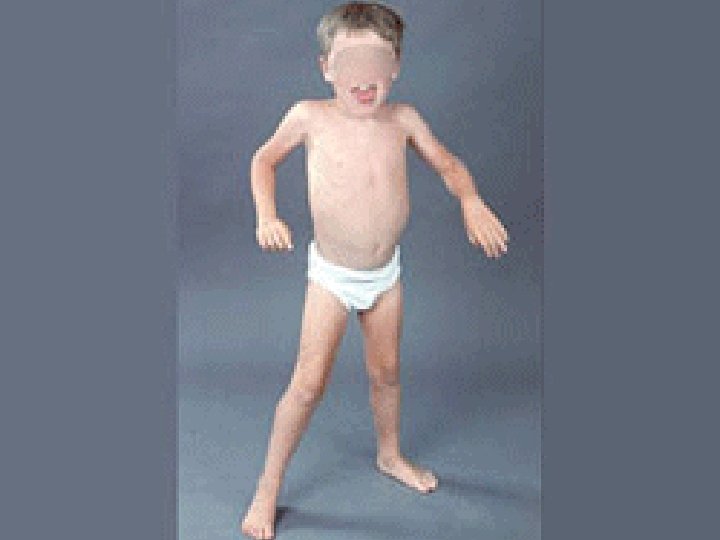
Clinical appearance 1. Muscle weakness. 2. Enlargement of calf muscles. Pseudohypertrophy: Muscle is replaced by adipose tissue & fibrosis Incresed muscle bulk.
3. Cardiac M. damage & fibrosis Heart failure & arrhythmias (fatal). 4. Due to ongoing muscle degeneration: High serum creatine kinase levels present at birth & persist through first decade of life but fall as muscle mass is lost during disease progression. It is important enzyme in skeletal muscles which consume ATP rapidly as it serves as energy reservoir for regeneration of ATP. (ATP can be generated gain).
Becker muscular dystrophy BMD: Becomes symptomatic later in childhood or adolescence. Progresses at slower rate. Many patients live well into adulthood & have a nearly normal life span. Cardiac involvement: Dominant clinical feature, result in death inspite of absence of significant skeletal muscle weakness.
Thank you