Multiple sequence alignment Why multiple sequence alignment The


















 is used to create pairwise alignments")



 operational")












- Slides: 37
Multiple sequence alignment
Why multiple sequence alignment • The DNA sequences of different organisms are often related (orthologos or paralogs? ) • Genes are conserved across widely divergent species, often performing a similar or even identical function, and at other times, mutating or rearranging to perform an altered function through the forces of natural selection. • Thus, many genes are represented in highly conserved forms in organisms. • Through simultaneous alignment of the sequences of these genes, sequence patterns that have been subject to alteration may be analyzed
Source: lecture by Serafim Batzoglou, http: //ai. stanford. edu/~serafim/CS 262_2009/index. php http: //www-personal. umich. edu/~lpt/fgfrcomp. htm
• As with aligning a pair of sequences, the difficulty in aligning a group of sequences varies considerably with sequence similarity. • If the amount of sequence variation is minimal, it is quite straightforward to align the sequences, even without the assistance of a computer program. • If the amount of sequence variation is great, it may be very difficult to find an optimal alignment of the sequences because so many combinations of substitutions, insertions, and deletions, each predicting a different alignment, are possible.
Challenges of the MSA • Finding an optimal alignment of more than two sequences that includes matches, mismatches, and gaps, and that takes into account the degree of variation in all of the sequences at the same time poses a very difficult challenge. • DP can be extended to three sequences, for more than three sequences, only a small number of relatively short sequences may be analyzed. Thus, approximate methods are used. • A second computational challenge is identifying a reasonable method of obtaining a cumulative score for the substitutions in the column of an MSA. • Finally, the placement and scoring of gaps in the various sequences of an msa presents an additional challenge.
Approximate methods • A progressive global alignment of the sequences starting with an alignment of the most alike sequences and then building an alignment by adding more sequences • Iterative methods that make an initial alignment of groups of sequences and then revise the alignment to achieve a more reasonable result • Alignments based on locally conserved patterns found in the same order in the sequences • Use of statistical methods and probabilistic models of the sequences
Use of MSA • A group of similar sequences may define a protein family that may share a common biochemical function or evolutionary origin. • Multiple sequence alignment of a set of sequences can provide information as to the most alike regions in the aligned sequences. In proteins, such regions may represent conserved functional or structural domains. • When dealing with a sequence of unknown function, the presence of similar domains in several similar sequences implies a similar biochemical function or structural fold that may become the basis of further experimental investigation.
Use of MSA • If the structure of one or more members of the alignment is known, it may be possible to predict which amino acids occupy the same spatial relationship in other proteins in the alignment. • In nucleic acids, such alignments also reveal structural and functional relationships. For example, aligned promoters of a set of similarly regulated genes may reveal consensus binding sites for regulatory proteins.
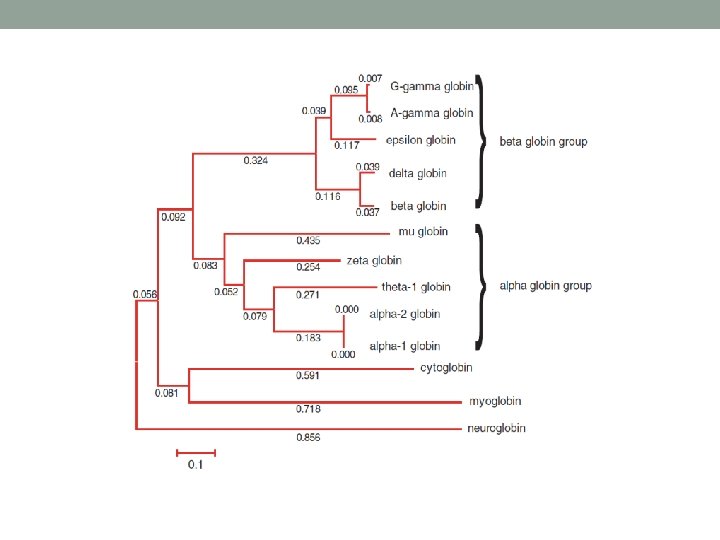
Use of MSA • Once the msa has been found, the number or types of changes in the aligned sequence residues may be used for a phylogenetic analysis. • A phylogenetic analysis of a family of related nucleic acid or protein sequences is a determination of how the family might have been derived during evolution. The evolutionary relationships among the sequences are depicted by placing the sequences as outer branches on a tree.
source: Pevsner, Bioinformatics and Functional Genomics
Source: NCBI
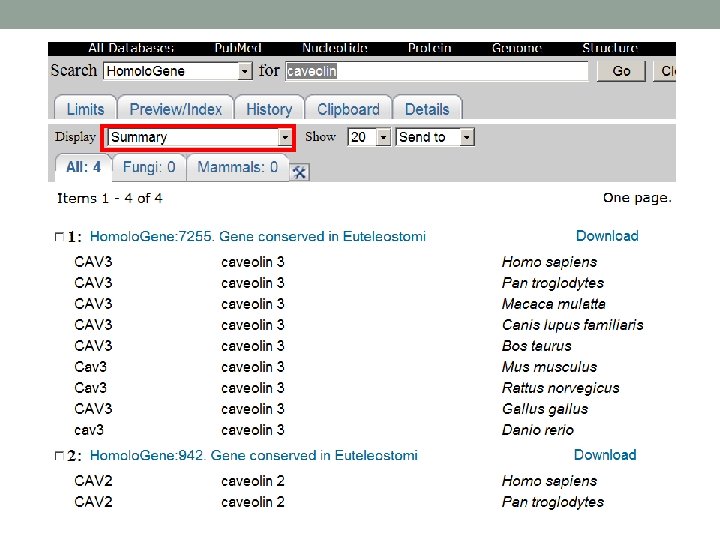
Example of MSA You are interested in Caveolin, search NCBI Homolo. Gene database
These nine proteins align nicely, although gaps must be included.
Progressive sequence alignment • The most commonly used algorithm, the most commonly used software Clustal. W • Popularized by Feng and Doolitle, often referred by these two names. • Permits the rapid alignment of even hundreds of sequences. • Limitation: the final alignment depends on the order in which sequences are joined. • Thus, it is not guaranteed to provide the most accurate alignments.
Clustal. W • http: //www. clustal. org • http: //www. ebi. ac. uk/Tools/msa/clustalw 2/ • EMBOSS – a free open source software analysis package (European Molecular Biology Open Software Suite) http: //emboss. sourceforge. net • Program emma is a Clustal. W wrapper • A variety of EMBOSS servers hosting emma are available, e. g. http: //embossgui. sourceforge. net/demo/emma. html • Clustal. X – a downloadable stand-alone program offering a graphical user interface for editing multiple sequence alignments • http: //www. clustal. org/clustal 2/
Clustal. W – how it works? • Three stages • Search hemoglobin subunit beta in NCBI Entrez
• Find similar sequences? Which tool would you use? • BLAST • Download msa 1. fasta • Perform the Clustal. W alignment at the EBI website
1 st stage • The global alignment (Needlman-Wunsch) is used to create pairwise alignments of every protein pair.
1 st stage • observed score for two of the two alignedaverage sequence i and j mean alignment score derived scores for the two from many (e. g. 1000) random sequences compared to shufflings of sequences i and j themselves
2 nd stage • A guide tree is calculated from the distance matrix. • The tree reflects the relatedness of all the proteins to be multiply aligned. Newick format
Guided tree • Guide trees are usually not considered true phylogenetic trees. • They are templates used in the third stage of Clustal. W to define the order in which sequences are added to a multiple alignment. • A guide tree is estimated from a distance matrix of the sequences you are aligning. • In contrast, a phylogenetic tree almost always includes a model to account for multiple substitutions that commonly occur at the position of aligned residues.
Construction of guided tree • Unweighted Pair Group Method with Arithmetic Mean (UPGMA) operational taxonomic units • A simple hierarchical clustering method • How it works? http: //www. southampton. ac. uk/~re 1 u 06/teaching/upgma/ • Neighbor joining • Uses distance method, distance matrix is an input. • The algorithm starts with a completely unresolved tree (its topology is a star network), and iterates over the following steps until the tree is completely resolved and all branch lengths are known.
Source: wikipedia
3 rd stage • The multiple sequence alignment is created in a series of steps based on the order presented in the guide tree. • First select the two most closely related sequences (see above) from the guide tree and creates a pairwise alignment. • The next sequence is either added to the pairwise alignment (to generate an aligned group of three sequences, sometimes called a profile) or used in another pairwise alignment. • At some point, profiles are aligned with profiles. • The alignment continues progressively until the root of the tree is reached, and all sequences have been aligned.
Gaps • “once a gap, always a gap” rule • The most closely related pair of sequences is aligned first. • As further sequences are added to the alignment, there are many ways that gaps could be included. • Gaps are often added to the first two (closest) sequences. • To change the initial gap choices later on would be to give more weight to distantly related sequences. • To maintain the initial gap choices is to trust that those gaps are most believable.
Iterative approaches • Compute a sub-optimal solution using a progressive alignment strategy and keep modifying that intelligently using dynamic programming or other methods until the solution converges. • i. e. they create an initial alignment and then modify it to try to improve it. • Progressive alignment methods have the inherent limitation that once an error occurs in the alignment process it cannot be corrected, and iterative approaches can overcome this limitation. • e. g. MUSCLE, Iter. Align, Praline, MAFFT
MUSCLE • Since its introduction in 2004, the MUSCLE program of Robert Edgar has become popular because of its accuracy and its exceptional speed, especially for multiple sequence alignments involving large numbers of sequences. • Multiple sequence comparison by log expectation • Three stages
1 st stage • A draft progressive alignment is generated. • Determine pairwise similarity through k-mer counting (not by alignment). • Compute distance (triangular distance) matrix. • Construct tree using UPGMA. • Construct draft progressive alignment following tree.
2 nd stage • Improve the progressive alignment. • Compute pairwise identity through current MSA using the fractional identity. • Construct new tree using Kimura distance matrix. • In a comparison of two sequences there is some likelihood that multiple amino acid (or nucleotide) substiutions occurred at any given position, and the Kimura distance matrix provides a model for such changes. • Compare new and old trees: if improved, repeat this step, if not improved, then we’re done.
3 rd stage • Refinement of the MSA • Systematically partition the tree to obtain subsets; an edge (branch) of the tree is deleted to create a bipartition. • Extract a pair of profiles (multiple sequence alignments), and realign them • Accept/reject the new alignment based on the sum-ofpairs score increase/decrease. • All edges of the tree are systematically visited and deleted to create bipartitions. • This iterative refinement step is rapid and had been shown earlier to increase the accuracy of the multiple sequence alignment.
Benchmarking the algorithms • Many algorithms exist, see e. g. http: //en. wikipedia. org/wiki/Sequence_alignment_software #Multiple_sequence_alignment • How can we assess the accuracy and performance properties of the various algorithms? • A convincing way to assess whether a multiple sequence alignment program produces a “correct” alignment is to compare the result with the alignment of known threedimensional structures as established by x-ray crystallography.
Benchmark data sets • Several databases have been constructed to serve as benchmark data sets. Database URL BAli. BASE http: //www-bio 3 d-igbmc. u-strasbg. fr/balibase/ IRMBASE http: //dialign-tx. gobics. de/main Ox. Bench http: //www. compbio. dundee. ac. uk/Software/Oxbench/oxbench. htm Prefab http: //www. drive 5. com/muscle/prefab. htm SABmark http: //bioinformatics. vub. ac. be/databases/content. html
• These are reference sets in which alignments are created from proteins having known structures. • Thus, one can study proteins that are by definition structurally homologous. This allows an assessment of how successfully assorted multiple sequence alignment algorithms are able to detect distant relationships among proteins. • For proteins sharing about 40% amino acid identity or more, most multiple sequence alignment programs produce closely similar results. • For more distantly related proteins, the programs can produce markedly different alignments, and benchmarks are useful to compare accuracy.
Benchmark metrics • Commonly used is sum-of-pair scores SPS. • This involves counting the number of pairs of aligned residues that occur in the target and reference alignment, divided by the total number of pairs of residues in the reference.