Multiple Sequence Alignment MultSeqAlign allows to detect similarities



 SP – the sum of pairwise scores of all")
")





 Scoring scheme – distance. (b) Scoring scheme satisfies the triangle")
 Find Sc minimizing")
 is at most twice the score of the D(Mopt) D (Mc) / D")
 k Σj=1. . k, j c D(Sc, Sj) = Σi=1. . k Σ")



 = Σ i con D(Scon , Si ) (a) Scon")
 Mj - column j of mult. align.")
 = Σi d(x*j, Mj) The optimal consensus multiple")
/k for: (a) SP mult. align. (b) Consensus")






 (a)Pick an unaligned")
 1. Calculate the pairwise")
 1. Calculate the pairwise")

 From the profile build")
- Slides: 34
Multiple Sequence Alignment • Mult-Seq-Align allows to detect similarities which cannot be detected with Pairwise-Seq-Align methods. • Detection of family characteristics. Three questions: 1. Scoring 2. Computation of Mult-Seq-Align. 3. Family representation.
Multiple Sequence Alignment
Scoring: SP (sum of pairs) SP – the sum of pairwise scores of all pairs of symbols in the column. (-, -) = 0 ρ3(-, A, A) = (-, A)+(A, A) SP Total Score = Σ ρi
Induced pairwise alignment or projection of a multiple alignment. a(S 1, S 2 ) a(S 2, S 3) a(S 1, S 3) SP Total Score = Σi<j score[ a(Si, Sj ) ] (-, -) = 0
Dyn. Prog. Solution
Dynamic Programming Solution • The best multiple alignment of r sequences is calculated using an r- dimensional hyper-cube • The size of the hyper-cube is O( Πni ) • Time complexity O(2 r nr) * O(computation of the ρ function). • Exact problem is NP-Complete (metrics: sum-of-pairs or evolutionary tree). more efficient solution is needed
Multiple Alignment from Pairwise Alignments ? Problem: • The best pairwise alignment does not necessary lead to the best multiple alignment.
Pattern-X Pattern-A Pattern-B S 1 Pattern-A Pattern-X Pattern-D S 2 Pattern-D Pattern-B Pattern-X S 3 S 1 S 2 S 1 S 3 Pattern-A Pattern-B Empty S 2 S 3 Pattern-D Correct Solution S 1 S 2 S 3 Pattern-X
S 1 S 2 S 3 ? ? ? ? Global or Local alignment?
Center Star Alignment (a) Scoring scheme – distance. (b) Scoring scheme satisfies the triangle inequality: for any character a, b, c dist(a, c) ≤ dist(a, b) + dist(b, c) (c) S 3 S 2 S 1 (in practice not all scoring matrices satisfy the triangle inequality) (d) (c) D(Si, Sj ) – score of the optimal pairwise alignment. (d) D(M) = Σi<j a. M (Si, Sj ) – score of the multiple alignment M. (e) a. Mc(Sc, Si) – pairwise alignment/score induced by Mc. Sc Sk Sk-1 Sk-2
S 3 S 2 The Center Star Algorithm: S 1 (a) Find Sc minimizing Σi c D(Sc , Si ). Sc (b) Iteratively construct the multiple alignment Mc: 1. Mc={Sc} Sk Sk-1 Sk-2 2. Add the sequences in S{Sc} to Mc one by one so that the induced alignment a. Mc(Sc, Si) of every newly added sequence Si with Sc is optimal. Add spaces, when needed, to all pre-aligned sequences. Running time: * O(n 2). AC-BC DCABC AC--BC DCA-BC DCAABC
D(Mc) is at most twice the score of the D(Mopt) D (Mc) / D (Mopt) ≤ 2(k-1)/k ( < 2 ) Proof: (a) a(Si, Sj) ≥ D (Si, Sj ) (any induced align. is not better than optimal align. ) a. Mc (Sc, Sj) = D (Sc, Sj ) (b) a. Mc (Si, Sj) ≤ a. Mc (Si, Sc) + a. Mc (Sc, Sj) = D (Si, Sc ) + D (Sc, Sj ) (follows from the triangle inequality) (c) 2 D(Mc) = Σi=1. . k Σ j=1. . k, j i a. Mc (Si , Sj ) ≤ Σi=1. . k Σ j=1. . k, j i ( a. Mc (Si, Sc) + a. Mc (Sc, Sj) ) 2(k-1) Σj c a. Mc (Sc, Sj) = 2(k-1) Σj c D(Sc, Sj) =
(d) k Σj=1. . k, j c D(Sc, Sj) = Σi=1. . k Σ j=1. . k, j c D(Sc, Sj) ≤ Σi=1. . k Σ j=1. . k, j i D(Si, Sj) ≤ Σi=1. . k Σ j=1. . k, j i a. Mopt (Si, Sj) = 2 D(Mopt) (e) → → 2 D(Mc) ≤ 2(k-1) Σj c D(Sc, Sj) k Σj c D(Sc, Sj) ≤ 2 D(Mopt) D(Mc)/(k-1) ≤ Σj c D(Sc, Si) ≤ 2 D(Mopt)/k → D (Mc) / D (Mopt) ≤ 2(k-1)/k
Scoring Metrics
Question: How to represent a family? • Consensus sequence • Profiles, HMM • Signature
Consensus Sequence Seq 1 -> Seq 2 -> Seq 3 -> Consensus-> aba ab– –ba aba
Consensus error: Notice: E(Scon) = Σ i con D(Scon , Si ) (a) Scon is not necessary one of the input strings. Steiner string S* : (b) Consensus error and Steiner string are defined without Mult. Align. E(S*) = min. S E(S) Approximation Algorithm: E(Scenter star)/E(S*) ≤ 2(k-1)/k (in case that scoring scheme satisfies the triangle inequality)
Mult. Align. -> Consensus Sequence Definitions: (a) Mj - column j of mult. align. M (b) Mi, j – character in row i, column j of mult. align. M (c) Distance between the character x and column j: d(x, Mj) = Σi dist(x, Mi, j) Given mult. align. M, the consensus character x* of column ‘j’ of mult. align. M is such that: d(x*, Mj) = minx d(x, Mj) Consensus string SM : SM = x*1 x*2 … x*q
Alignment Error of SM : σ(SM) = Σi d(x*j, Mj) The optimal consensus multiple alignment is a multiple alignment M with consensus string S*M such that : σ(S*M) = min M, SM σ(SM) It can be shown that: The optimal consensus multiple alignment specifies the optimal Steiner string S* and vice versa – from S* we can construct the optimal consensus multiple alignment.
Conclusion: Center Star method approximation error 2(k-1)/k for: (a) SP mult. align. (b) Consensus mult. align.
Profiles Seq 1 -> Seq 2 -> Seq 3 -> Seq 4 ->
Profile Analysis M. Gribskov, D. Eisenberg. Profile Analysis - detection of distantly related proteins by sequence comparison. The information is expressed in a positionspecific scoring table (profile).
Profile alignment • Sequence – Profile Alignment. • Profile – Profile Alignment. Dynamic Programming. (the same idea as in Pairwise Sequence Alignment)
reminder: Pairwise Sequence Alignment The position-specific gap Sequence-Profile alignment: coefficients penalize gaps in conserved regions more S(x, j) – aligning ‘x’ with column ‘j’ heavily than gaps in more S(x, j)= Σy σ(x, y) p(y, j)/p(x) variable regions σ(x, y) – any regular score for Pairwise Alignment (PAM-k, BLOSUM-k …) p(x, j) – frequency that character x appears in mult. align. column ‘j’ p(x) – frequency that character x appears anywhere in all sequences from mult. align.
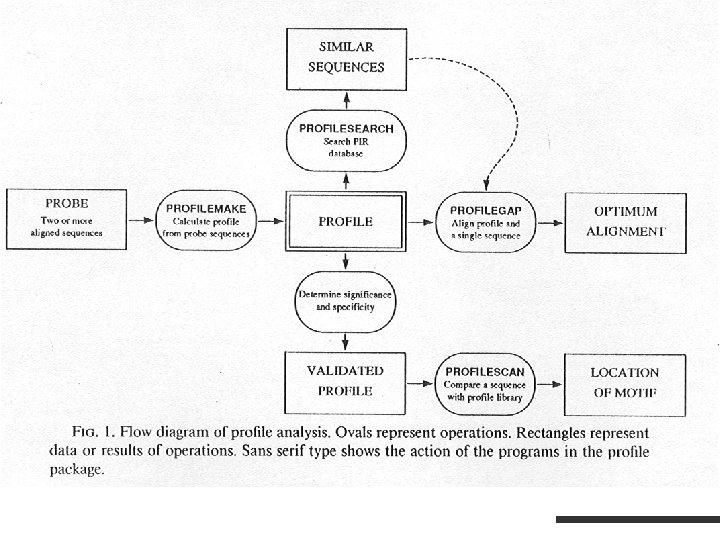
Profiles in GCG Pile. Up creates a multiple sequence alignment from a group of related sequences. Profile. Make makes a profile from a multiple sequence alignment. Profile. Search uses the profile to search a database for sequences with similarity to the group of aligned sequences. Profile. Segments displays optimal alignments between each sequence in the Profile. Search output list and the group of aligned sequences (represented by the profile consensus). Profile. Gap makes optimal alignments between one or more sequences and a group of aligned sequences represented as a profile. Profile. Scan uses a database of profiles to find structural and sequence motifs in protein sequences.
Iterative pairwise alignment 1. Align some pair. 2. While (not done) (a)Pick an unaligned string which is ”near” some aligned one(s). (b)Align with the profile of the previously aligned group. Resulting new spaces are inserted into all strings in the group.
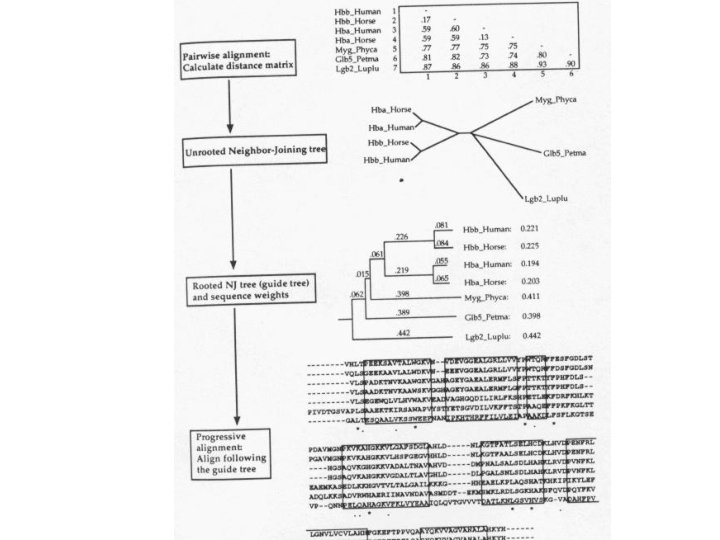
Progressive Alignment Feng-Doolittle 1987 Implemented in Pile. Up (GCG package) 1. Calculate the pairwise alignment scores, and convert them to distances. 2. Use an incremental clustering algorithm to construct a tree from the distances. 3. Traverse the nodes in their order of addition to the tree, progressively aligning the sequences. This way, the most similar pair is aligned first, followed by the addition of the next most similar sequence or set of sequences.
Progressive Alignment Clustal. W (algorithm of Thompson, Higgins, Gibson 1994) 1. Calculate the pairwise alignment scores, and convert them to distances. 2. Use a neighbor-joining algorithm to build a tree from the distances. 3. Align sequence - sequence, sequence - profile, profile - profile in decreasing similarity order.
Alignment tree built by Clustal. W
Profile HMMs Aligning a string S versus a profile: (a) From the profile build a HMM (b) Calculate a likelihood of S against the HMM Mi - main state, models the column i of the mult. align. Ii - insert state Di - delete state