MPZ mutations associated with deafness and abnormal pupillar

MPZ mutations associated with deafness and abnormal pupillar reaction in Czech CMT 2 patients, but also in HMSN III patients. P. Seeman 1, K. Huehne 2, R. Mazanec 3, B. Rautenstrauss 2, V. Beneš jr. 1, P. Šušlíková 1, O. Keller 4. 1 - Dept of Child Neurology, 3 - Dept of Neurology 2 nd School of Medicine, Charles University Prague, 2 - Institute of Human Genetics, University Erlangen, 4 - Dept of Neurology Thomayer University Hospital Prague.

Different HMSN phenotypes associated with MPZ mutations. • Charcot – Marie – Tooth type 1 B (demyelinating) • Dejerine – Sottas neuropathy (DSN) • Congenital hypomyelination neuropathy (CHN) • Charcot – Marie – Tooth type 2 (axonal) - „new“

Deafness in CMT • Deafness was reported in some of the CMT 2 families with MPZ mutations (Thr 124 Met) • Deafness was not observed in HMSN type III patients or families associated with MPZ mutations
.")
Myelin protein zero (MPZ).

Myelin protein zero gene. 173 b p exon 1 307 b p exon 2 278 b p exon 3 307 b p exon 4 309 b p exon 5 exon 6
. Recent findings")
Axonal - CMT 2 phenotype due to mutation in myelin gene (MPZ). Recent findings of MPZ mutations in CMT 2 patients and families with distinct phenotype (Ser 44 Phe, Asp 61 Gly, Asp 75 Val, Ile 99 Thr, Tyr 119 Cys, Thr 124 Met, Gln 141 stop, …) • • axonal type of CMT - EMG and nerve biopsy late onset polyneuropathy abnormal pupillar reaction – slow or absent deafness - usually after the onset of neuropathy

Czech family with axonal CMT beginning with deafness

Family F.

Family F. • hearing loss and deafness as the first symptom of CMT disease - at the late teens in the grandfather and in the mother - progressive hearing loss, deaffness now • abnormal pupillar reaction (Argyll-Robertson pupilles, Adie pupilles) before the onset of the neuropathy • fully normal fysical abilities untill the end of 3 rd decade • late onset of polyneuropathy of axonal type - slow progression at the grandfather but quite fast at the mother, 12 years old boy clinically still unaffected • severe distal muscle atrophies in mother and grandfather • whorsening of electrophysiological findings correlated with higher age in the family – axonal loss and later
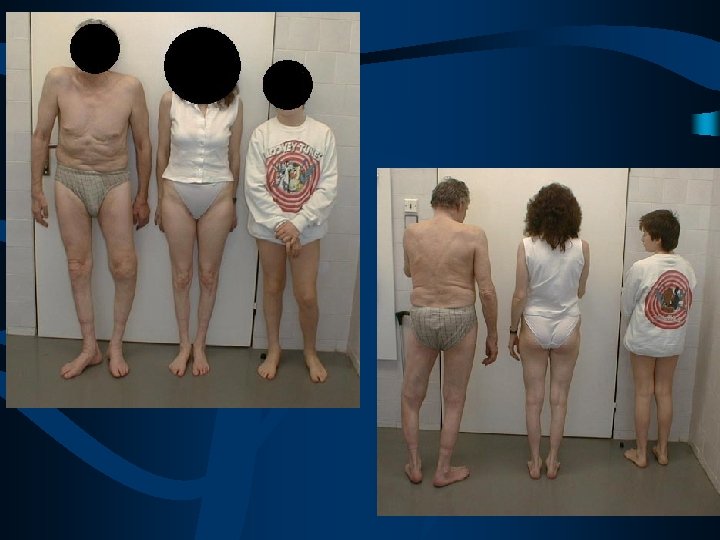

distal atrophies and weakness

no pes cavus, severe footdrop pronounced hand muscles atrophies
 findings I. Motor and sensory nerve conduction velocities Patient 645")
Electrophysiological (EMG, VEP, BAEP) findings I. Motor and sensory nerve conduction velocities Patient 645 Tibial Grandf. Median Motor nerve DML CMAP (ms) (m. V) none 9. 4 0. 1 29 Sensory nerve SCV SNAP (m/s) (u. V) Sural none Median none 14 7 47 47 16 26 MCV (m/s) 622 moth) Tibial Median none 4. 8 0. 4 30 Sural Median 623 boy) Tibial Median 6. 2 3. 5 10. 3 13. 2 40 45 Sural Median
 findings II. Brainstem auditory evoked potentials (monoaural, rarefaction click, 10")
Electrophysiological (EMG, VEP, BAEP) findings II. Brainstem auditory evoked potentials (monoaural, rarefaction click, 10 Hz ) Patient Side Latencies of waves /ms/ 645 G L R I III no response V I-III III-V 622 M L R 2. 36 2. 30 4. 44 4. 32 6. 29 none 2. 08 2. 02 1. 85 none 3. 93 none 623 B L R 1. 68 1. 98 3. 92 4. 15 5. 58 6. 11 2. 24 2. 17 1. 66 1. 96 3. 90 4. 13 Upper limits of normal range of BAEPs in our laboratory / +- 2 SD /: Latencies : I 2. 04 ms III 4. 16 ms V 6. 06 ms I – III 2. 16 ms III – V 2. 04 ms I – V 4. 15 ms I-V
 findings III. Visually evoked potentials (pattern reversed, monocular, 2 Hz,")
Electrophysiological (EMG, VEP, BAEP) findings III. Visually evoked potentials (pattern reversed, monocular, 2 Hz, 16 ´´) N 75 Patient 645 Side Left Right 622 L R 623 L R 79 79 Latencies of waves / ms / P 100 N 145 104 105 136 137 68 67 100 127 124 76 76 124 116 189 193 Limits of normal range of VEPs in our laboratory / +- 2 SD / Latencies : N 75 / 67 – 84 ms / P 100 / 94 – 116 ms / N 145 / 125 – 155 ms / Amplitude: N 75 / P 100 5. 4 u. V
 VEP – normal latencies BAEP – prolonged latencies")
Results from the mother (nr. 622) VEP – normal latencies BAEP – prolonged latencies - peripheral Audiometry – profound perception hearing loss brain MRI – normal, only diffuse nonspecific white matter changes in both hemispheres CSF – normal protein and normal cell count muscle biopsy – neurogenic lesion, fiber hypertrophy and atrophy nerve biopsy – done in 1993, axonal loss, axonal lesion
 in MPZ gene")
290 A>T (Glu 97 Val) in MPZ gene
 nerve muscle Normal control")
Nerve and muscle biopsy in nr. 622 (mother) nerve muscle Normal control
")
MRI in nr. 622 (mother)
")
MRI in nr. 622 (mother)

Dejerine Sottas neuropathy and MPZ mutation • Deafness was usually not reported in DSS patients – (but the reported patients were usually children or young persons, few data about adult DSS patiens and their hearing status)

Family K. • Mutation Arg 98 Cys neighbour aminoacid to the previous family
 and son (18 y. ) severely affected")
Family K. • Mother (44 y. ) and son (18 y. ) severely affected (HMSNIII or DSN), no other affected members in the family • early onset (3 y. ) with hypotonia, delayed motor milestones, scoliosis, both affected never achieved normal independent gait • distal weakness and atrophies, areflexia, rather nonprogressive course • extremely decreased MNCV ( 8 -10 m/s), absent SNAP • CSF - ? • hypertrofic demyelinating neuropathy (with onion- bulbs) in the sural nerve biopsy in the mother • in the mother from the age of 25 y. - hearing loss, presently sensorineural hearing loss bilateral ( up to 70 d. B) both affected showed abnormal pupillar reaction - extremely prolonged opening reaction (opening)

Family K. mother son

Family K. mother son

Conclusions • Is deafness a common feature associated with MPZ mutations, but later in life (in adult age) ? • Do also other HMSN III or DSN patients develop hearing loss in adulthood ? • In our CMT 2 family F. (Glu 97 Val), deafness was by far the first symptom of the disease – before any signs of HMSN • Testing of the functional effect of the newly discovered mutations with CMT 2 phenotype in expression systems should clarify the mechanism leading to axonal demage resulting from some MPZ mutation • Only the location in the extracellular domain alone, do not explain really different phenotypes resulting from mutation of the neibour aminoacids ( Lys 96 Glu – CMT 1, Glu 97 Val – CMT 2, Arg 98 Cys – DSN, Arg 98 His - Ile 99 Thr – CMT 2) some other mechanism is more probable.
- Slides: 26