Motor Neuron diseases Classification Lower motor neurons disorders




")


 Lower Motor neuron Disorders")

 Proximal autosomal recessive SMA of childhood Type. I: Werdnig hoffman disease: -")









 is involved in transport of m-RNA in the")













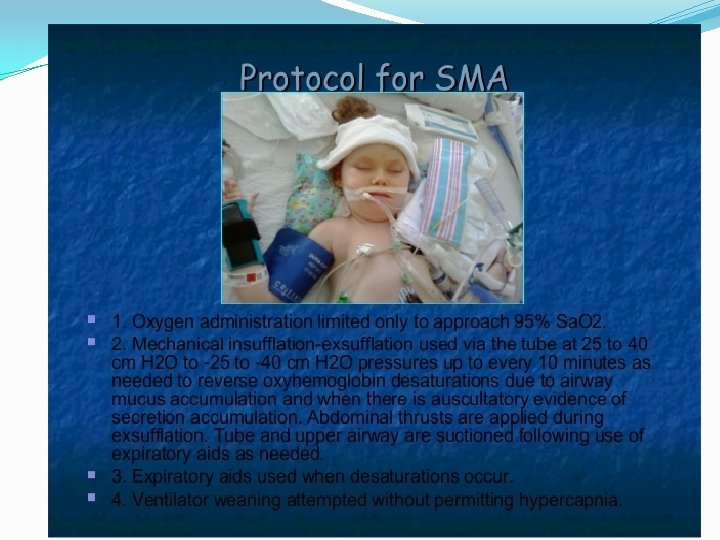








 � 90%")












 �Stage 1: Mild weakness, But pt is ambulatory independent �Rx:")
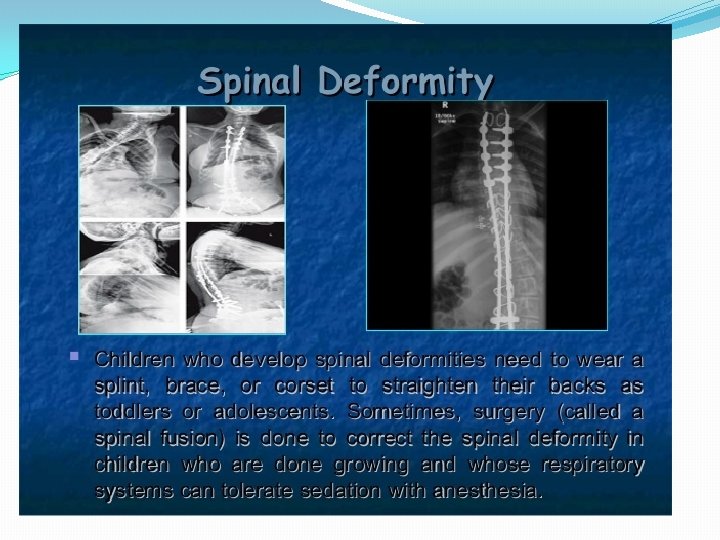
 �Stage 4: Shoulder subluxation, edema, �Techniques to relieve")

- Slides: 67
Motor Neuron diseases
Classification Lower motor neurons disorders: �-- Hereditary : Spinal Muscular Atrophy �-- Acquired: i. Monomelic focal and segmental spinal muscular atrophies ii. Multifocal motor neuropathies iii. Acute motor axonal neuropathy iv. Post polio syndrome v. Post irradiation syndrome � --Infective disorders
Combined upper and lower motor neuron disorders: Amyotrophic Lateral sclerosis: i. Familial Adult onset ii. Familial juvenile onset iii. Sporadic iv. ALS plus syndromes like… ALS and frontotemporal dementia, Western Pacific ALS-Parkinsonismdementia complex.
Upper Motor Neuron disorders -- Primary Lateral sclerosis -- The hereditary spastic paraplegias -- Neurolathyrism -- Konzo
�Disorders of the bulbar motor system -- Kennedy’s disease ( X linked bulbospinal neuropathy) -- Brown- Vialetto –van Laere syndrome -- Fazio-londe disease
�Toxic disorders of the motor neuron -- Neurolathyrism --Heavy metal toxicity -- Post irradiation motor neuron injury -- Western Pacific ALS –Parkinsonism-dementia complex
�Disorders of motor neuron over activity -- Neuromyotonia -- Stiff person syndrome �Miscellaneous motor neuron disorders -- Endocrinopathies -- Copper deficiency syndrome -- Benign cramp fasciculation syndrome
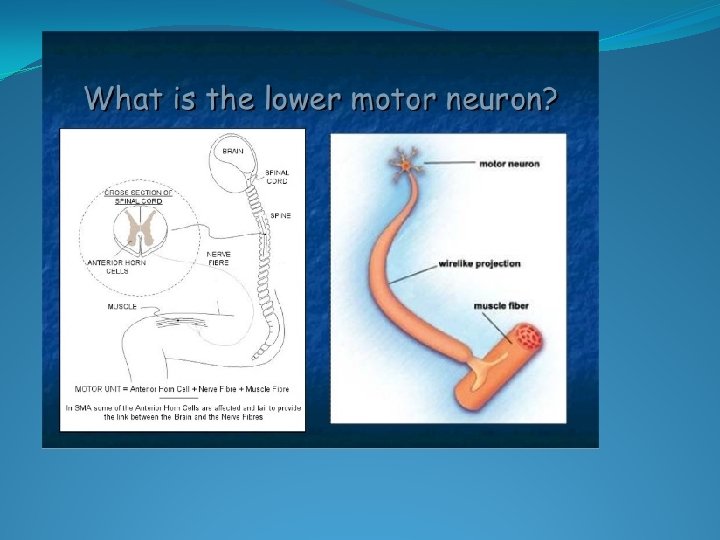
I) Lower Motor neuron Disorders
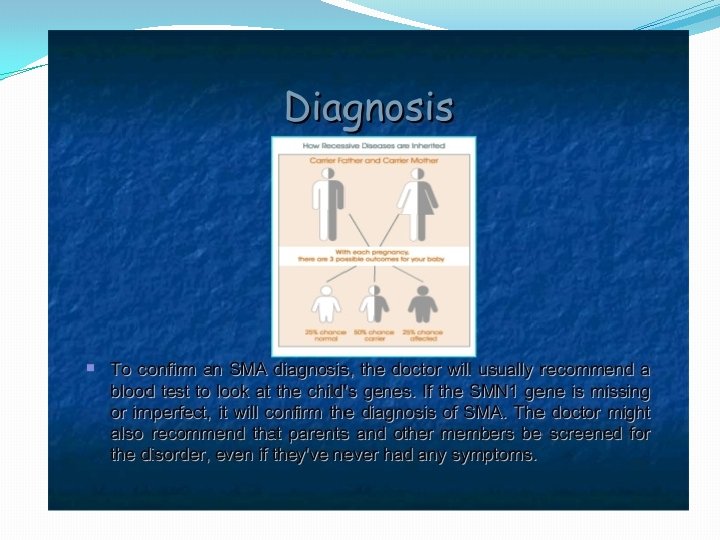
�Hereditary: Spinal muscular atrophy: A group of genetically determined pure lower motor neuron disorders in which degeration of the anterior horn cells leads to progressive, symmetrical muscle weakness and wasting with sparring of sensations and absence of pyramidal tract affection.
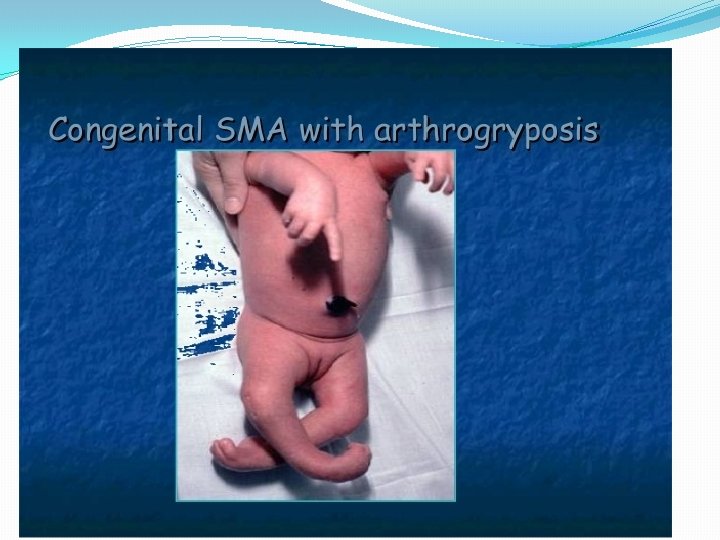
� 1) Proximal autosomal recessive SMA of childhood Type. I: Werdnig hoffman disease: - Presents with severe generalised weakness, hypotonia at birth or by 6 months of age - Affected children never sit or walk - Usually die of respiratory insufficiency within 2 years - Type 2: Intermediate form: onset of muscle weakness before 18 months of age - -- patients can sit but cannot walk unaided. - -- survival limited to adolescence
�Type 3: Kugelberg- Welander disease: -- Onset of weakness after 18 months -- usually are able to stand walk -- may become wheelchair dependant by adulthood -- life expentancy is normal �Type 4: Adult onset : is designated as type 4
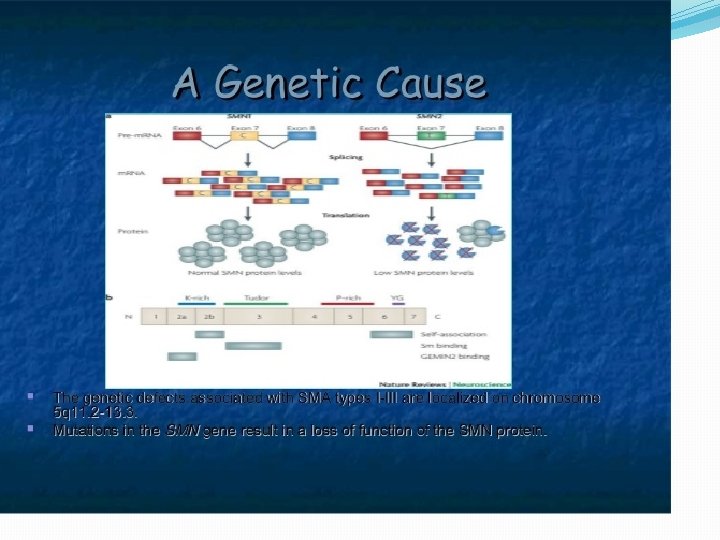
�SMN ( survival motor neuron gene) is involved in transport of m-RNA in the axons of motor neurons. In an event of abnormality of the gene the protein synthesis required for the structural continuity of the axons is hampered.
� A few variants of SMA: -- Sinal muscular atrophy with pontocerebellar hypoplasia: Additional features are cortical blindness, nystagmus, mental retardation -- Autosomal recessive Distal SMA: Slow progressive involvement of distal extremities with variable age of onset. Here there may be upper limb, lower limb or vocal cord predominance.
�In SMARD there may be slight sensory and autonomic nervous system involvement
�Congenital nonprogressive spinal muscular atrophy affecting the lower limbs �Scapuloperoneal SMA: -- muscular weakness in scapuloperoneal distribution -- May be myogenic and neurogenic in origin -- There have been evidences for neuronal degeneration, pure myopathis and combinations of pathologies.
Drugs commonly used �Neuroprotective agents: inhibition of endogenous cell death pathways eg: Gabapentine, Riluzole -- Thyrotropin releasing hormone: neurotrophic effects on the anterior horn cells �Anabolic agents like albuterol have been used: improvement in muscle strength has been observed �Myostatin inhibitor: Myostatin is a negative regulator of satellite cells thus comes in the way of mitosis.
�Muscle strengthening �Muscle extensibility �Orthotic care �Enviornmental modification �Maintaining respiratory hygiene �Vocational training
Amyotrophic Lateral Sclerosis
�Also called the Lou Gehrig’s disease �Major loss of both Upper and Lower motor neurons --- Anterior horn cells in spinal cord --- Motor cranial nerve neuclei in Brainstem leading to Amyotrophy ---- Demyelination and Gliosis of Corticospinal and Corticobulbar tracts due to degeneration of Betz cells in the Motor cortex … leading to lateral sclerosis.
Epidemiology �Mean age of onset: 57 years (50 -70 years of age) � 90% cases are sporadic, � 10% may be familial
Etiology Suggested �Toxicity due to abnormal levels of Magnesium, Copper, alluminium has been suggested �Deficiency of nerve growth factor �Viral origin �Mutation of Superoxide Dismutase gene on chromosome 21 in familial forms of ALS, leading to increase in toxic properties �Oxidative stress and aberration in the exitotoxicity pathways leading to cell death
Diagnosis �EMG: fibrillations, fasciculations seen in the muscles at rest and large unit spikes with voluntary activity �In 70% of patients: elevated levels of Creatinine Phosphokinase �CSF and blood tests used only to rule out other conditions �Superoxide Dysmutase gene for the familial forms �NCS: normal distal conduction velocity, F wave abnormalities �MRI: Wallerian degeneration in Corticospinal and corticobulbar tracts
Differential diagnosis �Multiple sclerosis �Cervical Myolopathy �Syringomyelia �Peripheral neuropathies �Other motor neuron diseases
Clinical Features �Weakness and atrophy: in all 4 limbs, mostly progressing from distal to proximal �Fatigue, cramps �By the time the patient comes with complaints, motor neuron degeneration would have progressed �Bulbar symptoms occur later in the disease �Sensory and ANS symptoms absent…. . Have been positive in a few cases with sensory evoked potential abnormalities picked up.
�Bulbar symptoms include: tongue faciculations, facial and palatal weakness, . . Dysphagia and dysarthria �Occulomotor neuclei are almost always spared �Death is usually due to respiratory failure �Amyotrophic Lateral Sclerosis severity scale: -- Lower extremity: Walking -- Upper Extremity: ( Dressing and Hygiene) -- Speech -- Swallowing
Prognosis �Average survival post onset is 4 years. �A small number have lived for 15 to 20 years post onset. �Patients with initial involvement of respiratory and the bulbar muscles are known to have faster progression of the disease. �Prognosis may vary as drug therapies are developed.
Medical Management Drugs under investigations include: -- Gabapentin: to decrease the synthesis of glutamate -- Supplemental dosage of Tocopherol: Vit E an antioxidant and free radical scavenger -- Insulin like growth factor (rh. IGF-I) -- Riluzole: decreases the presynaptic release of Glutamate has been found to have positive effects on the length of survival
Supportive therapy for clinical problems �Muscle relaxants for spasms and cramps like Quinine and Baclofen. �For dysphagia: Problems with management of their saliva. . Chocking, drooling, increased viscocity due to dehydration -- Hydration, tablets containing papain and bromelain. -- Anticholinergic agents -- sometimes surgical options like ligation of ducts, severing the parasympathetic supply to the salivary glands, botox, radiotherapy
�Dysarthria: Palatal lift prosthesis to address hypernasality, abnormalities with volume and speed of speech, -- speech therapy, breathing techniques. -- Voice amplification systems -- Home made point boards -- Computer based communication based on single pointing, eye movements that translates written word into voiced words
�Respiratory Management: -- Weakness of diaphragm, intercostals, abdominals, other accessory muscles. -- signs are orthopneoa, hypoventilation, weak ineffective cough -- O 2 saturation levels ( transcutaneously measured) -- Forced Vital capacity assessment in standing and supine. Rx: Chest Pt: Breathing for energy conservation, Stretching to maintain the length of muscles, endurance and strength traning, -- Oxygenation at 2 L/min -- Bi PAP at home -- Home mechanical ventilation ( tracheostomy may be needed
Physiotherapy management �Daily activity log : -- Type of activity -- What position are you in ( Lying, sitting, standing, moving) -- Fatigue level ( 0 -10) -- Pain ( Location, intensity from 0 to 10) �Assessment of muscle tone, strength, endurance, tightness, respiratory function, pain, speech �Contextual factors
�Principles to be followed 1. Combine formal exercises with enjoyable physical activities 2. Activities with oppurtunities for social development and personal accomplishment 3. Strengthening for concentric rather than eccentric activities as eccentric can cause muscle damage which in dennervated muscles may not be reversed 4. Mderate resistence exs programs as high resistance donot have an added advantage 5. Focus on stengthening stronger muscles as those with < antigravity strength have less chance of improving 6. Monitor muscle power for overwork weakness 7. Activity modifications should include periods of physical activity and rest.
Phase I ( Independent) �Stage 1: Mild weakness, But pt is ambulatory independent �Rx: Prevent disuse atrophy Add strengthening, ROM exs. �Stage 2: Moderate selective weakness �Rx: Avoid contractures, orthotic support, Strengthening with caution, use adaptive equipment to facilitate ADLs �Stage 3: Severe selective weakness, increased respiratory effort, easy fatiguability �Rx: Chest PT, Wheelchair: modified for the patient, Head rest may be needed. , add pleasurable activities.
Phase 2: ( Partially independent) �Stage 4: Shoulder subluxation, edema, �Techniques to relieve pain, spasm, shoulder stability, over head slings for transitions �Stage 5: Wheelchair dependance complete, secondary skin changes, sever UL and LL weakness. �Techniques for tranfers taught to care givers, HMV, Antipressure mattresses, home modifications
Phase III: Dependant �Bed ridden �Rx: For dysphagia: long spoons, feeding tubes, gastrotomy �Suctioning �Speech amplification, eye pointing �Clearing of airways, tracheostomy care