Molecular Simulation Molecular Simluation Introduction Prerequisition A powerful





















: • Born-Oppenheimer approx.")









- Slides: 39
Molecular Simulation
Molecular Simluation • Introduction: • Prerequisition: • A powerful computer, fast graphics card, • An efficient way to solve Schroedingers wave equation for many atoms, • A reliable structure or structural model
Molecular Simluation • Structures or reliable model structures: • X-ray crystallography • NMR spectroscopy • Homology modelling
Molecular Simulation • Where do we get the structures from? • Databases • Brookhaven protein data base • Or homology models
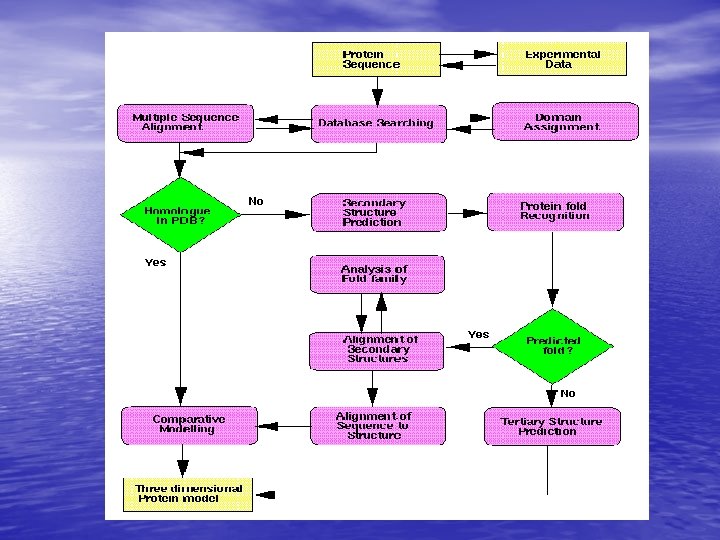
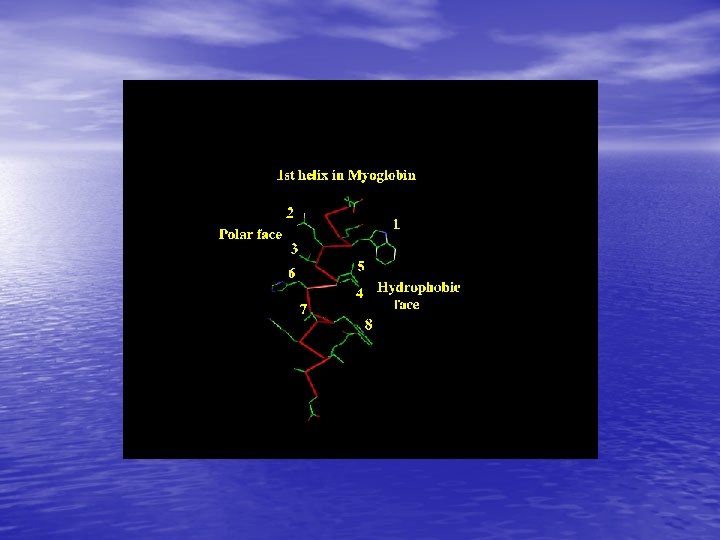
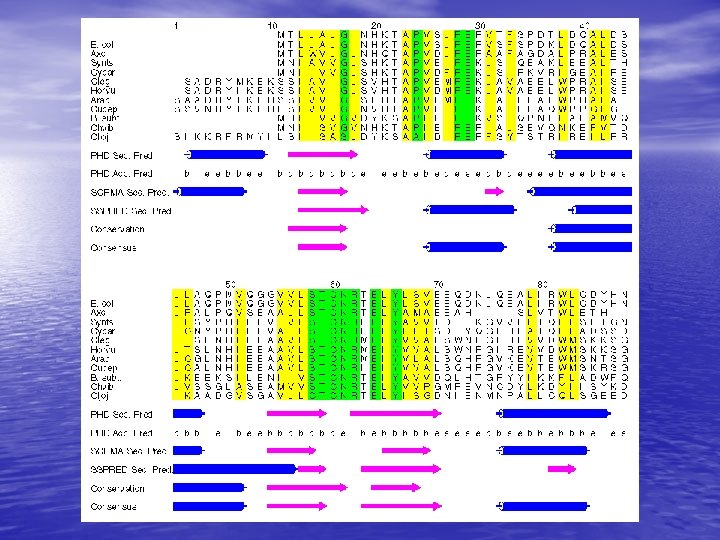
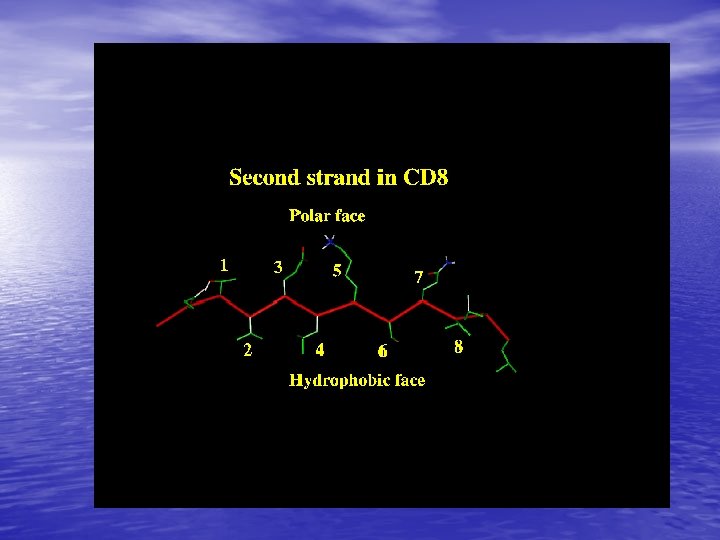
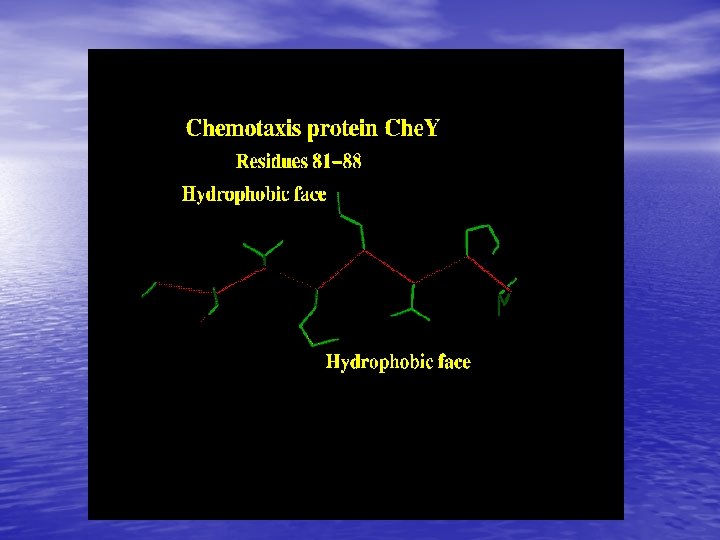
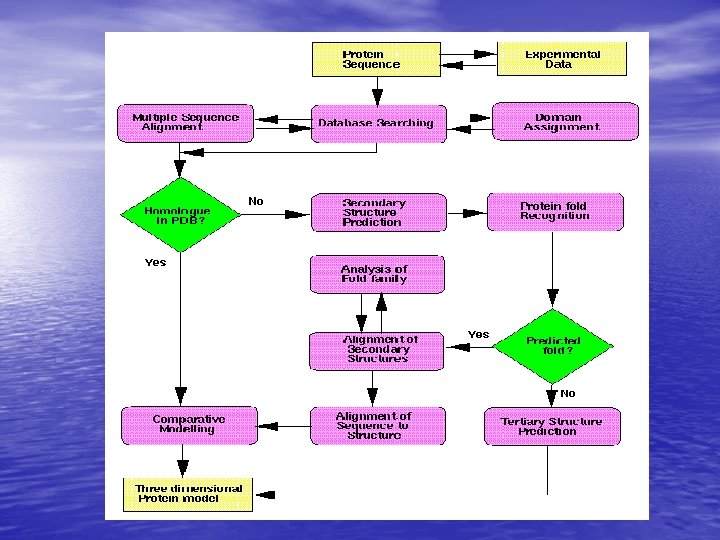
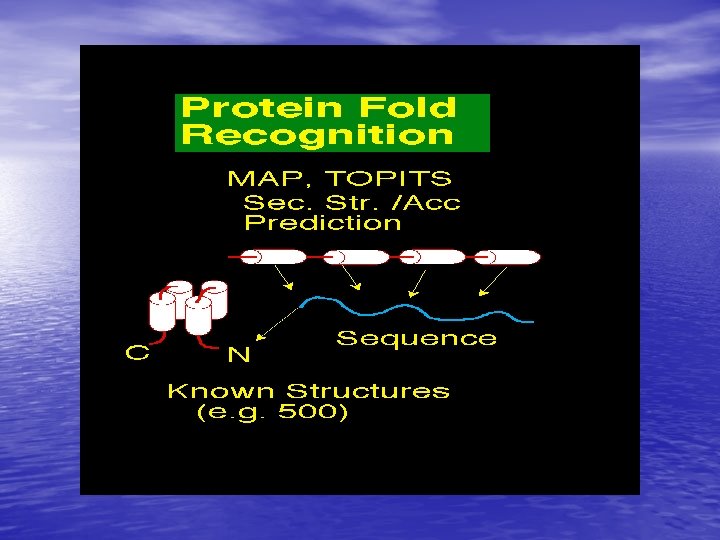
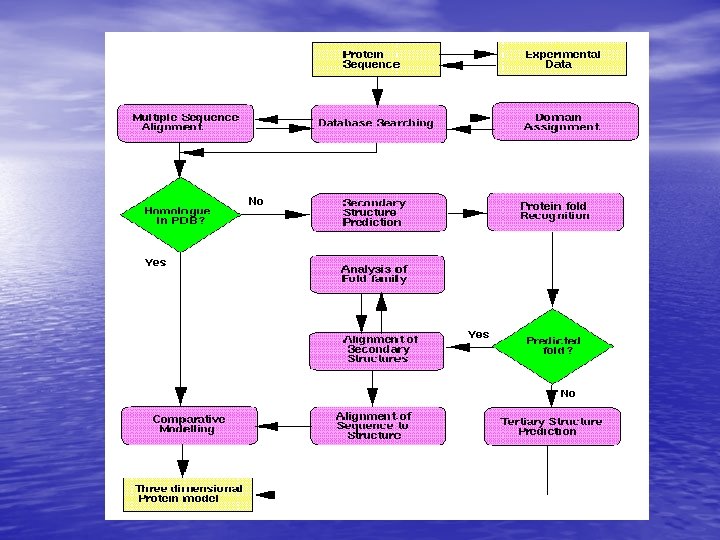
Molecular Simulation • Homology modelling:
Molecular Simulation • Molecular Mechanics: • The Potential energy surface (PES): • Born-Oppenheimer approx. allows us to separate • • • electronic and nuclear motion when nuclei move, the electrons re-adjust quickly energy of molecule is a function of the positions of the nuclei potential energy surface describes energy of a molecule in terms of its structure
Molecular Simulation
Molecular Simulation • • • Structure determined from the potential energy surface minimum corresponds to an equilibrium structure first order saddle point corresponds to a transition state for a reaction path is the steepest descent path connecting a transition state to minima • • • Dynamics molecules move on the potential energy surface dynamics can be treated classically or quantum mechanically small amplitude motions - normal modes of vibration large amplitude motions - trajectories, wave packets, reactions statistical mechanics to connect microscopic to macroscopic
Molecular Simulation • Bond stretching • Valence angle bending • Torsions • Van der Waals Interactions • Electrostatic Interactions • Cross terms
Molecular Simulation • Bond Stretching and valence angle bending
Molecular Simulations • Torsions
Molecular Simulation • Interaction of 2 point charges
Molecular Simulation • Van der Waals Interactions
Molecular Simulation • Force Field Energies • Geometry Optimisation • Thermodynamics
Molecular Simulation • Modern Force Fields • • CHARMM Biomolecules CHARMm Bio and org. molecules CFF/CVFF org. and bio molecules GROMOS Biomolecules MM 2 organics MM 3 org. and bio molecules OPLS all-atom and united atom version SYBYL/TRIPOS org. and bio molecules