Mol Mov DB Database of molecular movements Anastasia

Mol. Mov. DB: Database of molecular movements Anastasia Kurdia

Motivation Motion is closely related to the way a structure fulfills a particular function, and protein motion is involved in wide variety of basic protein functions Studying motions provides an insight into function
")
Motivation Rapidly increasing amount of data characterizing protein structure (X-ray crystallography, NMR, computational data) Classification of motions helps in predicting protein function
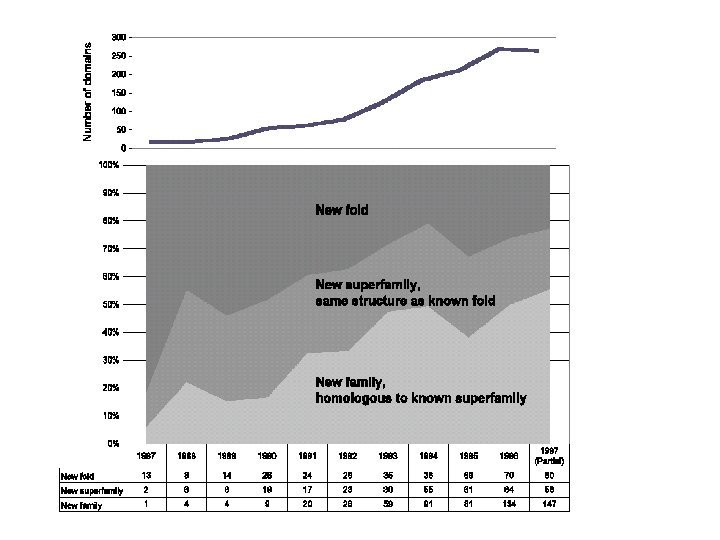

Motivation • • Need for data storage and management Need for an efficient mechanism to relate the structures one to another, beyond classification based on sequence or structural alignment.

Mol. Mov database • • • Storing motions Obtaining motions Classification of motions Standardized statistics and quantitative measures Unified nomenclature for protein motions Movies as database entries Custom software tool to perform service tasks – morph server
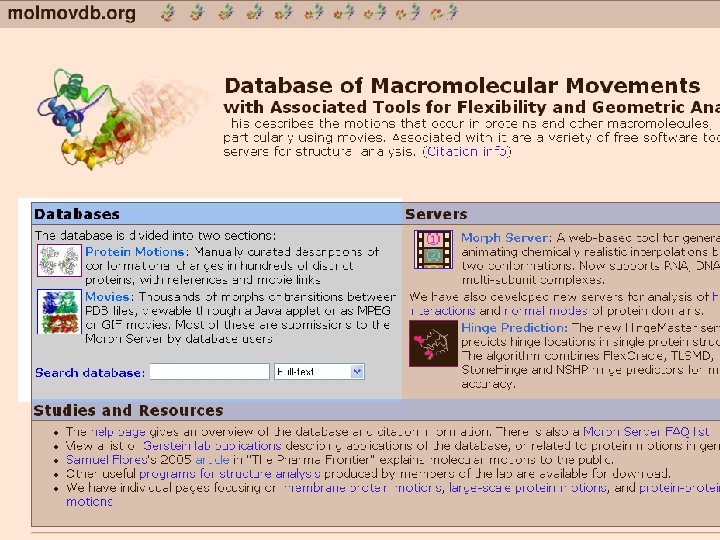

Protein motions Manually maintained descriptions of conformational changes in hundreds of distinct proteins, with references and movie links.
![Possible ways a motion can be classified [D-s-2] Known Domain Motion, Shear Mechanism [D-h-2]](http://slidetodoc.com/presentation_image_h/333d40b26b384b9a301375d08e8f38af/image-9.jpg "Possible ways a motion can be classified [D-s-2] Known Domain Motion, Shear Mechanism [D-h-2]")
Possible ways a motion can be classified [D-s-2] Known Domain Motion, Shear Mechanism [D-h-2] Known Domain Motion, Hinge Mechanism [D-? -2] Known Domain Motion, Unclassifiable Mechanism [D-n-2] Known Domain Motion, Neither Hinge nor Shear Mechanism [D-f-2] Known Domain Motion, Partial Refolding of Structure [D-s-1] Suspected Domain Motion, Shear Mechanism [D-h-1] Suspected Domain Motion, Hinge Mechanism [D-? -1] Suspected Domain Motion, Unclassifiable Mechanism [D-n-1] Suspected Domain Motion, Neither Hinge nor Shear Mechanism [F-s-2] Known Fragment Motion, Shear Mechanism [F-h-2] Known Fragment Motion, Hinge Mechanism [F-? -2] Known Fragment Motion, Unclassifiable [F-n-2] Known Fragment Motion, Neither Hinge nor Shear Mechanism

Sample entry in Protein motions • • http: //www. molmovdb. org/cgibin/motion. cgi? ID=calbind&graphics=1

Movies Thousands of morphs of transitions between PDB files, viewable through a Java applet or as MPEG or GIF movies. Most of these are submissions to the Morph Server by database users

Sample entries in Movies • • http: //www. molmovdb. org/cgibin/morph-classic. cgi? ID=34318 -16829 http: //www. molmovdb. org/cgibin/morph. cgi? ID=63984 -22780

Structural domain A structural domain of a protein is a selfstabilizing structural element that often folds independently of the rest of the protein chain. Many domains are not unique to the proteins produced by one gene or one gene family but instead appear in a variety of proteins

Hinge vs. shear motion Hinge motion is characterized by large changes in main-chain dihedral angles occurring at a localized region (a hinge). Hinge motions are similar to rotations around an articulated joint and therefore can be very large. They usually involve a small number of residues, since even one bond can provide the required rotational freedom. • Shear motions are very limited and involve large number of residues.

Example of hinge motion: calmoduline Large-scale movement of calmodulin involving splitting of one long helix. The total rotation of one domain relative to the other is upwards of 150 degrees.

Morph server Issues Morph server steps Get start and end configuration Searches for homolog pairs Make them compatible Homogenizes input PDB files into a consistent format Find flexible regions Uses homogenized structures to perform a number of additional analyses (hinge location) Obtain motion Uses adiabatic mapping to interpolate PDB files Visualize output Allows visualization of interpolated structures in a variety of movie formats Quantitatively measure output Calculates statistics

Getting start and end configuration Automated search in SCOP, PDB, etc >1200 user submitted morphs Submitted Manual >14000 automatically classified motions Automatic As of 2003 ~200 manually classified motions

Making input homogenous • • Most of the truly novel functionality of the server Establish equivalence of sequences: alignment (AMPS for similar proteins, structural alignment for distant proteins)

Finding hinges • Construct a search window of 24 residues and examine each position along peptide backbone in the window. If one half in the window belongs to one domain and the other part belongs to another domain, a hinge is detected. • If hinges are not found along the backbone, a window is reduced by two and a scan is repeated. After a scan was performed 5 times and no hinges were detected, a failure is reported.

Finding hinges A stand-alone application Hinge master implements several techniques to efficiently spot hinges. find hinges access results

Interpolation: adiabatic mapping The morph server attempts to describe protein motions as a rigidbody rotation of a small “core” relative to a larger one, using a set of hinges. To ensure all statistics between any two motions are directly comparable, the motion is placed in a standardized coordinate system. A pathway interpolation is produced by two principal methods: • Straight Cartesian interpolation. The difference in each atomic coordinate (between the known endpoint structures) is calculated and then divided into a number of evenly spaced steps. • Adiabatic mapping. Energy minimization is added after each interpolation step. This procedure produces interpolated frames with much more realistic geometry.

Visual rendering Interpolation produces a set of. pdb files that can be used to produce a custom movie A movie is created on-the-fly: • a PDF file containing individual frames • Multi. Gif, Quicktime, MPEG with 2 D movie • a VRML 3 D movie (requires a plugin)

Statistics • • Maximum C alpha displacement, rotation angle in degrees around putative hinge regions and other parameters The statistics are detailed enough to perform automatic preliminary classification of the motion


New multichain morph server • • http: //molmovdb. org/cgi-bin/beta. cgi FRODA lite option added, allowing to invoke directed dynamics with default parameters Hydrogen bonds are not considered, adding protons is not required Paths produced by FRODA avoid sterically impossible trajectories
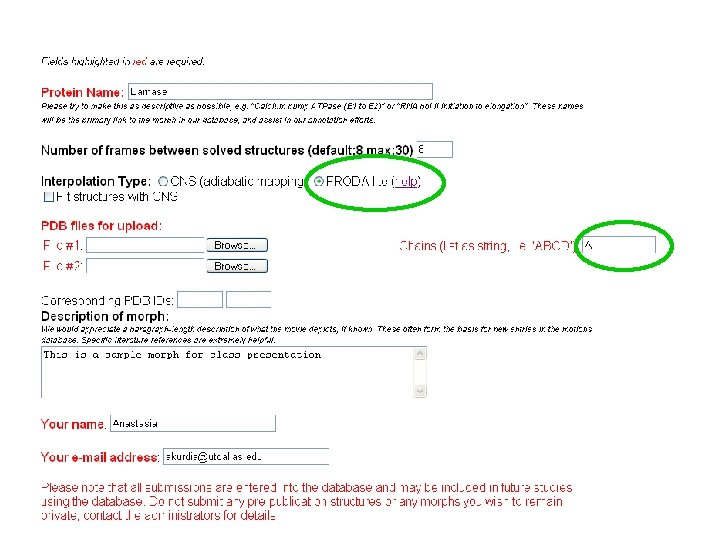

Sample morph Initial file and final result of FRODA simulation from previous class were fed to beta server directed motion of barnase

Alternatives • Rigi. Mol: • • Identifies rigid or semi-rigid structural domains. Displays RMSD, rotation, and translation statistics for the domains. Generates interpolated coordinates that effectively "morph" between the structures. Outputs standard PDB files to be used for visualization • LSQMAN: Structural alignment program that does simple morphing using Cartesian or "internal" coordinates. • Indie server at the NIH. Similar to the Morph Server, but uses linear interpolation and requires the submitted files to have the same sequence. • Dyn. Dom Performs detailed structural comparisons similar to the statistics from morph server. This has a database of motions as well.

Questionable elements • • Every user is free to enter data to Mol. Mov. DB The system cannot accommodate all types of motions Quality of a morph Quantitative measure

References • The Database of Macromolecular Motions: new features added at the decade mark. S Flores, N Echols, D Milburn, B Hespenheide, K Keating, J Lu, S Wells, EZ Yu, M Thorpe, M Gerstein (2006) Nucleic Acids Res 34: D 296 -301. • The morph server: a standardized system for analyzing and visualizing macromolecular motions in a database framework. WG Krebs, M Gerstein (2000) Nucleic Acids Res 28: 1665 -75. • Wells S. , Menor S. , Hespenheide B. M and Thorpe M. F. (2005) Constrained Geometric Simulation of Diffusive Motion in Proteins. Physical Biology 2, S 127 -S 136. • The Yale Morph Server (http: //molmovdb. org) • http: //www. public. asu. edu/~frasch/molecular_simulations. htm
- Slides: 30