MIASTENIA I ZESPOY MIASTENICZNE Prof dr hab med

MIASTENIA I ZESPOŁY MIASTENICZNE Prof. dr hab. med. Anna Kamińska Dr med. Anna Kostera-Pruszczyk

Miastenia i zespoły miasteniczne • choroby złącza nerwowo-mięśniowego • objawy: osłabienie i męczliwość mięśni poprzecznie prążkowanych • wrodzone, autoimmunologiczne, toksyczne i in.

Hughes i wsp. 2005

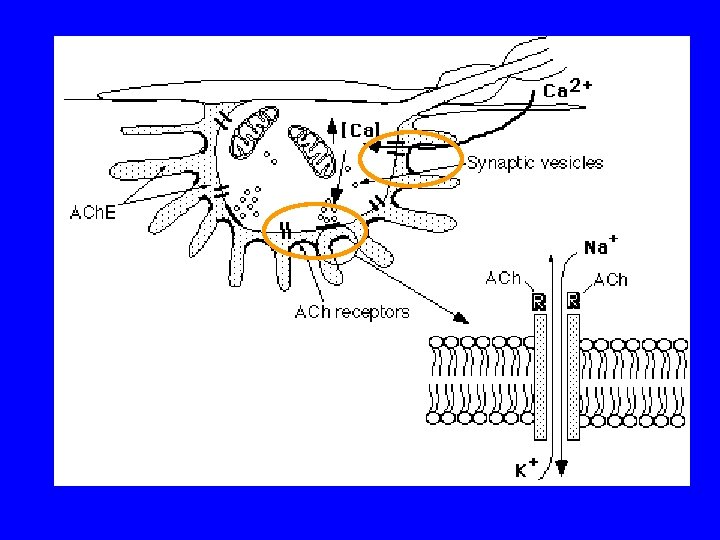
Receptor acetylocholinowy to kanał jonowy bramkowany ligandem Dimitru -95 Electrodiagnostic Medicine
 równomiernie rozłożony wzdłuż błony mięśniowej z gęstością")
• Mięsień niedojrzały: ACh. R (płodowy) równomiernie rozłożony wzdłuż błony mięśniowej z gęstością 1000/mm 2 (Bevan, Steinbach 1977) • dojrzała synapsa: gęstość ACh. R >10 000 /mm 2, ok. 10/mm 2 poza synapsą (Merlie, Sanes 1985)

• receptor płodowy: podjednostki 2 a, b, d, g • receptor dojrzały: podjednostki 2 a, b, d, e • typ płodowy po urodzeniu: – mięśnie oczne, część opuszkowych (Kaminski i wsp. 1995), – mięsień odnerwiony – reekspresja u pacjentów z niektórymi wrodzonymi zespołami miastenicznymi (Engel i wsp. 1996)

A. Pestronk, www. neuro. wustl. edu/neuromuscular/

Mięśnie oczne: • ekspresja typu płodowego ACh. R • unerwienie wieloaksonalne A. Mięsień gałkoruchowy myszy B. Przepona myszy Hughes i wsp. 2005 • duża precyzja ruchów - mała tolerancja na zaburzenia funkcji (Kaminski i wsp. 2003)
 • potencjał")
• miniaturowy potencjał płytki MEPP (uwalniane w spoczynku pojedyncze kwanty ACh) • potencjał czynnościowy synapsy EPP (suma wielu MEPP powstałych w wyniku synchronicznego uwolnienia dużej liczby kwantów ACh)

• EPP zależy od wielkości kwantów ACh, ich liczby oraz odpowiedzi ze strony receptora ACh • jeśli EPP osiągnie wartość progową depolaryzacja skurcz komórki mięśniowej • różnica między EPP a potencjałem progowym to tzw. współczynnik bezpieczeństwa synapsy W NORMIE KAŻDY EPP JEST PONADPROGOWY

EPP, AP
 •")
Miastenia: krótka historia • 1879 Erb, 1887 Oppenheim, 1893 Goldflam, Jolly 1899 (MG) • 1935 Mary Walker: neostygmina łagodzi objawy MG • 1973 Daniel Drachman: liczba receptorów ACh obniżona do 11 -30% normy • 1976 Jon Lindstrom: 85% pacjentów z MG ma przeciwciała przeciwko receptorowi ACh

MG: naturalny przebieg choroby • samoistna remisja w pierwszym roku choroby 12 -21% pacjentów (Oosterhuis 1989) • okres aktywnej choroby (narastania objawów) 3 -7 lat (Oosterhuis 1989, Grob 1999) • objawy oczne uogólniają się u 40 -50% pacjentów; uogólnienie zwykle w ciągu pierwszych 2 lat choroby

Miastenia • autoimmunologiczna vs. genetycznie uwarunkowany zespół miasteniczny • serologia • patologia grasicy: – grasiczak – przetrwała grasica

Rola ACh. RAb w patogenezie miastenii Wg A. Pestronka

Rola ACh. RAb w patogenezie miastenii Przeciwciała przeciwko receptorowi acetylocholiny u 85 -90% pacjentów z miastenią

Miastenia seronegatywna • ok. 10 -15% pacjentów bez przeciwciał przeciwko receptorowi acetylocholiny • poprawa po plazmaferezie, IVIg, możliwe bierne przeniesienie choroby • więcej zachowanych ACh. R niż w seropozytywnej MG • stopień zaburzeń transmisji nerwowomięśniowej podobny

Miastenia seronegatywna • surowice 60 -70% pacjentów z „seronegatywną” MG: przeciwciała anty-Mu. SK (Hoch 2001) • miastenia Mu. SK-pozytywna 0 -40% populacji pacjentów seronegatywnych (Vincent 2005) • Mu. SK (muscle specific tyrosine kinase) udział w tworzeniu skupisk ACh. R w błonie postsynaptycznej • częściej oporne na leczenie objawy opuszkowe (zanik mięśni) • MG z anty-Mu. SK: bez patologii grasicy • TYMEKTOMIA ? ? ?

Miastenia seronegatywna • surowice 60 -70% pacjentów z „seronegatywną” MG: przeciwciała anty-Mu. SK (Hoch 2001) • Mu. SK (muscle specific tyrosine kinase) udział w tworzeniu skupisk ACh. R w błonie postsynaptycznej • miastenia Mu. SK-pozytywna 0 -40% populacji pacjentów seronegatywnych (Vincent 2005) • częściej oporne na leczenie objawy opuszkowe (zanik mięśni) • MG z anty-Mu. SK: bez patologii grasicy • TYMEKTOMIA ? ? ?
 Wiek zachorowania K: M Hoch i wsp.")
Autor/populacja % Mu. SK pozytywnych (liczba dodatnich/badanych) Wiek zachorowania K: M Hoch i wsp. 2001 70% (n=17/24) Romi i wsp. 2005/Norwegia 0% Niks i wsp/Holandia 2007 Uwagi 2 -68 3: 1 Ciężka uogólniona MG 22% (n=5/23) 36% (n=35/97) Heterogenna etnicznie 2 -76 3: 2 Uogólniona Zhou i wsp. 2004/USA 40% 5 -60 2: 1 Uogólniona, 70% poprawa, 3 immunosupresja Ohta i wsp. 2004 /Japonia, Kobe 41% Jiian Horng i wsp. /Chiny 2004 3, 8% (n=1/26) 67 M Uogólniona, RF Lee i wsp. 2006/Korea 26% (n=4/11) 18 -57 4: 0 Tylko uogólniona, poprawa 100% Padua i wsp. 2006/Włochy 48% (n=25/52) 37, 3 SD 18, 9 5: 1 Uogólniona, 52% niewydolność oddechowa A. Kostera-Pruszczyk i wsp 2008 /Polska 6, 4% (n=4/62) 8, 7% uogólnionych 22 -40 3: 1 Uogólniona, Poprawa 100% Również w MG seropozytywnej*** Sprostowanie 2005

Miastenia bez grasiczaka; wiek zachorowania 906 pacjentów wg Sanders i wsp, 1997

MG: objawy kliniczne zmienna męczliwość mięśni, nasilana przez wysiłek, zmniejszająca się po wypoczynku

• objawy oczne: – dwojenie – ograniczenie ruchomości gałek ocznych – opadanie powiek (asymetria) – fotofobia Jako pierwsze lub jedyne objawy miastenii !!!

• objawy opuszkowe: – niewyraźna, nosowa mowa, zacichanie mowy – chrypka – zaburzenia żucia – zaburzenia połykania (płyny, szczególnie ciepłe) – jeśli utrudnione połykanie śliny konieczne żywienie przez sondę • osłabienie mięśni oddechowych
 • postać dwuobręczowa MG 2 -3%")
• osłabienie mięśni kończyn (ksobne>odsiebne) • postać dwuobręczowa MG 2 -3%

miastenia dziecięca • Ok. 10% pacjentów z miastenią <16 r. ż. • poniżej 10 r. ż. K=M, powyżej 10 r. ż. K>M • przeciwciała przeciwko receptorowi acetylocholiny ok. 50 -80% • seronegatywna MG vs. wrodzone zespoły miasteniczne

Miastenia dorosłych Grasiczak: ok. 10 -15% pacjentów z MG między 2045 r. ż. (MG ok. 30% pacjentów z grasiczakiem) ACh. RAb 95 -100% przetrwała grasica 60 -80% pacjentów

Miastenia przejsciowa noworodków • Przemijające objawy u dziecka urodzonego przez kobietę z MG • Bierne przekazanie przeciwciał prze łożysko • Samoograniczający się charakter • Dzieci około 15% matek (Kostera. Pruszczyk i wsp. 2005)

klasyfikacja kliniczna MG • Osserman I: objawy oczne II: uogólnione (A łagodne, B średnio nasilone) III: ciężkie uogólnione IV: późna ciężka MG V: zaniki mięśni
 • • • Klasa I: oczna Klasa II- łagodna")
klasyfikacja kliniczna wg MGFA (2002) • • • Klasa I: oczna Klasa II- łagodna Klasa III- o średnim nasileniu Klasa IV- ciężka Klasa V- intubacja
 • • • Klasa I: oczna Klasa II- łagodna")
klasyfikacja kliniczna wg MGFA (2002) • • • Klasa I: oczna Klasa II- łagodna Klasa III- o średnim nasileniu Klasa IV- ciężka Klasa V- intubacja a. kończynowe/osiowe b. opuszkowe/oddechowe

klasyfikacja kliniczna MG • Osserman I: objawy oczne II: uogólnione (A łagodne, B średnio nasilone) III: ciężkie uogólnione IV: późna ciężka MG V: zaniki mięśni • wg skali Amerykańskiego Towarzystwa Neurologicznego

MG: diagnostyka • Test z tensilonem • badania elektrofizjologiczne: elektrostymulacyjna próba męczliwości, elektromiografia pojedynczego włókna mięśniowego (SFEMG) • przeciwciała przeciwko ACh. R, Mu. SK • TK śródpiersia
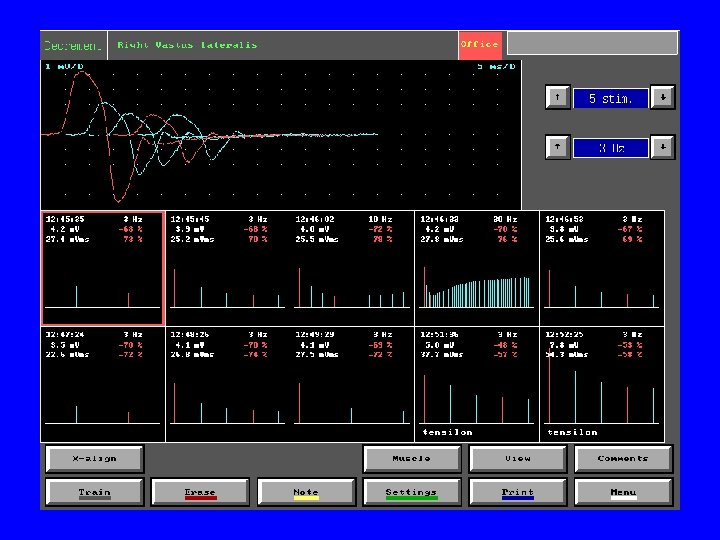

Wg Oh

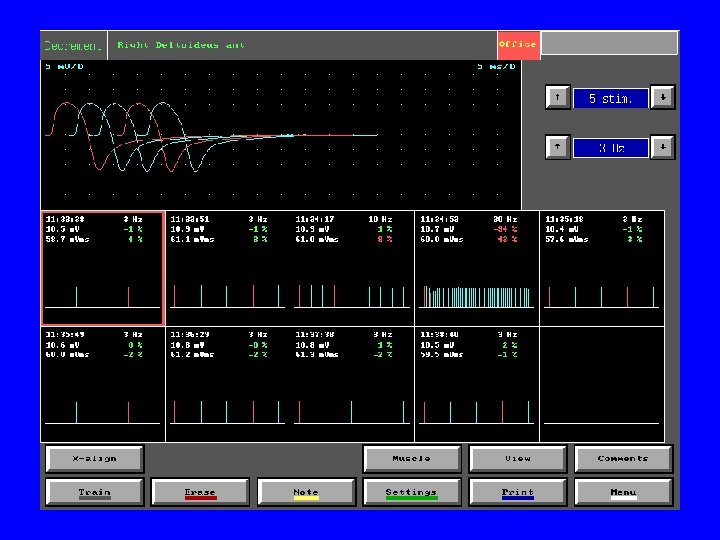
Single Fiber EMG Wyzwolenie Jitter Trigger 1 2 1 2 100 ms • 4 - 5 wkłuć w tym samym mięśniu • niewielki skurcz dowolny • Minimum 20 potencjałów 1 ms Jitter

zdrowy ochotnik oczna MG MG


MG Mu. SK-pozytywna bez patologii grasicy

MG: diagnostyka różnicowa • • • LEMS zatrucie toksyną botulinową PEO, miopatia mitochondrialna zapalenie wielomięśniowe kurcz powiek wrodzone zespoły miasteniczne
")
(wg Keesey 2004)

wg Grob, 1999

")
ZESPÓŁ LAMBERTA-EATONA patofizjologia • Blok kanałów Ca++ zwykle P/Q, rzadko N (przeciwciała Ig. G) • zmniejszone uwalnianie kwantów acetylocholiny z zakończeń presynaptycznych (prawidłowa liczba receptorów Ach)
, • w 90%")
ZESPÓŁ LAMBERTA-EATONA • osłabienie i męczliwość mięśni kończyn, (głównie mięśni ksobnych), • w 90% brak odruchów głębokich, • dysfunkcja układu autonomicznego (charakterystyczna suchość w ustach) • 85% pacjentów ma przeciwciała przeciwko kanałom Ca P/Q (Motomura 1995, Lennon 1995)
, białaczki, chłoniaki, rak")
ZESPÓŁ LAMBERTA-EATONA • w 60% współistnieje rak drobnokomórkowy płuc (O’Neil 1988), białaczki, chłoniaki, rak prostaty, ale również grasiczak!!! • LEMS może wyprzedzać kliniczne objawy choroby nowotworowej
: – obniżona amplituda odpowiedzi M – dekrement")
ZESPÓŁ LAMBERTA-EATONA • Obraz elektrofizjologiczny (Lambert 1965): – obniżona amplituda odpowiedzi M – dekrement przy stymulacji o niskiej częstotliwości – wzrost amplitudy odpowiedzi M przy stymulacji o wysokiej częstotliwości lub po 1015 sekundowym maksymalnym skurczu dowolnym

Zaburzenia transmisji nerwowomięśniowej w LEMS • tzw. triada Lambertowska • u pacjentów z LEMS torowanie istotne gdy przekracza 50% (O’Neil 1988), o min. 100% u 90% pacjentów (Maddison 1998)
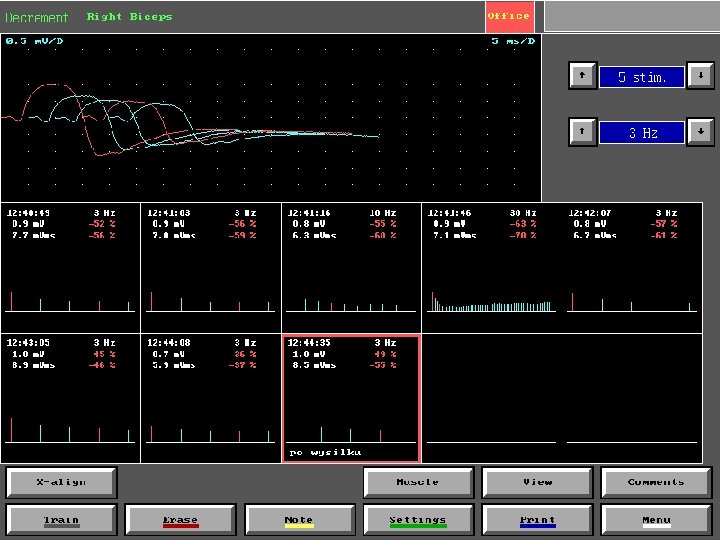
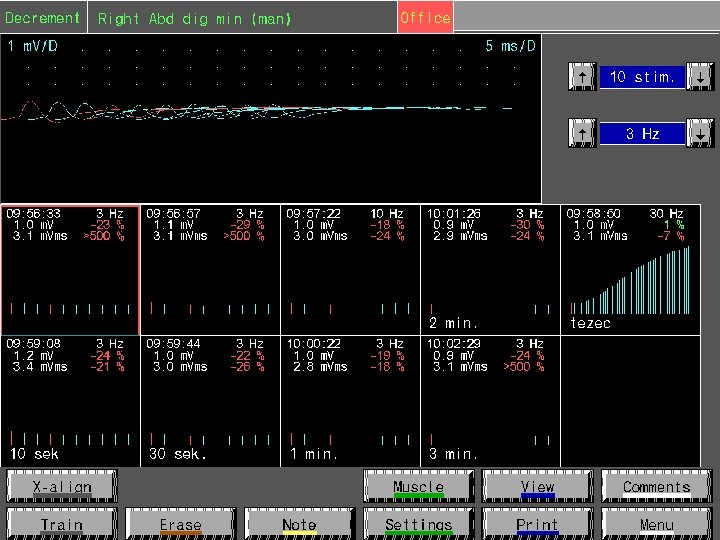

• Próba męczliwości zalecana jako pierwsze badanie diagnostyczne w przypadku podejrzenia MG: pomocna w różnicowaniu MG z LEMS • SFEMG może być wykonywane z pominięciem próby męczliwości tylko u pacjentów z oczną postacią MG • Prawidłowy wynik SFEMG w słabym klinicznie mięśniu wyklucza MG

WRODZONE ZESPOŁY MIASTENICZNE • rzadka i heterogenna grupa genetycznie uwarunkowanych zaburzeń złącza nerwowo -mięśniowego • klinicznie niedowład z męczliwością (fatiguing weakness) • początek we wczesnym dzieciństwie, ale czasem nawet w 6. dekadzie życia (A. Engel 2000)

WRODZONE ZESPOŁY MIASTENICZNE • diagnostyka różnicowa zależnie od wieku pacjenta, • fenotyp może przypominać zarówno MG jak i miopatie, neuropatie, choroby motoneuronu (SMA, ALS) (Engel at al. 2000)

Hantai i wsp. 2004

WRODZONE ZESPOŁY MIASTENICZNE • diagnostyka: – obraz kliniczny, ew. rodzinność – dodatnia elektrostymulacyjna próba męczliwości (czasem charakterystyczne cechy) • decydujące: – badania potencjałów wewnątrzkomórkowych in vitro – ocena morfologii płytki nerwowo-mięśniowej – badania genetyczne

Zespół wolnego kanału receptora acetylocholiny • najczęstszy WZM • pierwsze objawy kliniczne nawet w 6. dekadzie życia • objawy oczne i opuszkowe rzadko • objawy kończynowe, często wybitnie zajęte prostowniki palców i nadgarstka

Zespół wolnego kanału receptora acetylocholiny • dziedziczenie AD (opisano 15 różnych mutacji podjednostek ACh. R) • MEPP wydłużony, o obniżonej amplitudzie (wydłużony czas otwarcia kanału, kanał pobudzany również przez cholinę)

ZESPÓŁ WOLNEGO KANAŁU ACETYLOCHOLINY • w badaniu elektrofizjologicznym: – spadek amplitudy M przy stymulacji o częstotliwości 2 -3 Hz – w odpowiedzi na pojedynczy bodziec powtarzające się odpowiedzi M (repetitive), podanie Tensilonu nasila to zjawisko, zmniejsza stopień dekrementu UWAGA: podobny obraz daje również zespół synaptyczny: niedobór esterazy acetylocholinowej, tu Tensilon bez znaczenia
")
R. G. wrodzony zespół miasteniczny (wrodzony zespół wolnego kanału acetylocholiny)

Zatrucie toksyną botulinową • Clostridium botulinum: beztlenowiec • 7 typów neurotoksyny: A , B , C 1 , D , E, F, G • infekcje pokarmowe, przyranne, uogólnienie po miejscowym podaniu toksyny

Mechanizm działania toksyny botulinowej na złącze n-m • wiązanie z błoną presynaptyczną • internalizacja (aktywna endocytoza do zakończenia presynaptycznego • zahamowanie uwalniania ACh (hydrolizowanie białek: synaptobrewina-2, syntaksyna, SNAP-25) tzw. „chemiczna denerwacja” • reinerwacja rozpoczyna się ok. 7 dni później (nowe wypustki aksonalne)

Zatrucie toksyną botulinową • klinicznie: niedowład zwykle uogólniony, z przewagą ksobnych grup mięśni, objawy opuszkowe (dysfagia, dyzartria), zaburzenia gałkoruchowe, opadanie powiek, zaburzenia autonomiczne • w infekcjach pokarmowych czasem też objawy z przewodu pokarmowego (bóle brzucha, wymioty)

Strategia badania • wykazać zaburzenia transmisji nerwowomięśniowej oraz neurogenne cechy EMG w mięśniach • prawidłowe przewodzenie impulsów we włóknach czuciowych i ruchowych nerwów obwodowych (może być obniżona amplituda odpowiedzi M)

Badania elektrofizjologiczne w botulizmie • obniżona amplituda odpowiedzi M • w 50% przypadków dekrement przy stymulacji o częstotliwości 3 Hz, w 50% przypadków torowanie przy stymulacji o częstotliwości 50 Hz • w zapisie emg czasem małe potencjały j. r. • zawsze zaburzenia transmisji nerwowomięśniowej w SFEMG

SFEMG w zatruciu toksyną botulinową • jitter nieprawidłowy, wartości maksymalne w ciągu pierwszych 2 tygodni choroby • FD zawsze podwyższony, gdy jitter wydłużony • poprawie klinicznej towarzyszy stopniowa normalizacja wartości jitteru i równoległy wzrost wskaźnika FD (Olney, 1988)

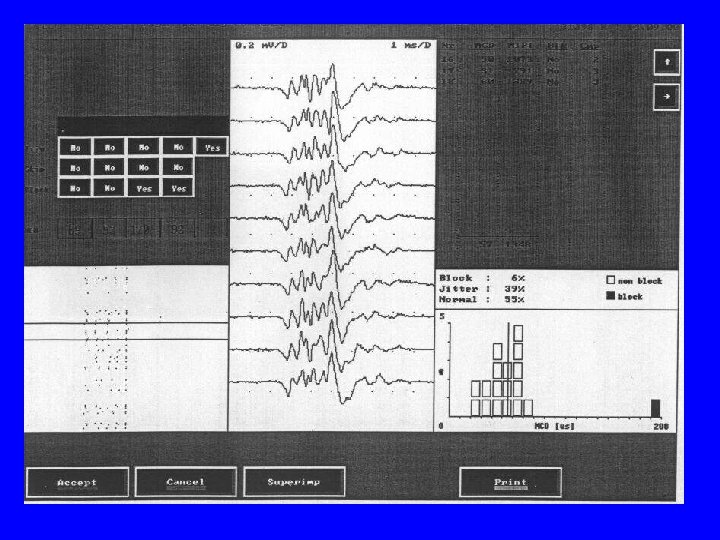

Leczenie • podtrzymujące, czasem konieczna wentylacja mechaniczna • Antytoksyna: – skuteczna w czasie pierwszej doby – trójwalentna ABE – decyzja o podaniu w oparciu o rozpoznanie kliniczne, nie czekając na potwierdzenie laboratoryjne

 2. Immunosupresja 3.")
LECZENIE MIASTENII 1. Poprawa transmisji nerwowomięśniowej, leczenie objawowe (inhibitory ACh. E) 2. Immunosupresja 3. Leczenie chirurgiczne (tymektomia) 4. Eliminacja krążących przeciwciał (plazmafereza-PE) 5. Leczenie immunomodulacyjne (Iv. Ig)

Inhibitory acetylocholinesterazy • leki objawowe • nie wpływają na aktywność procesu immunologicznego Początek działania Pirydostygmina (Mestinon) Ambenomium (Mytelase) Neostygmina (Polstygmina) Czas działania ok. 4 - 6 h po ok. ½- 1 h (p. o) po ok. ½- 1 h ok. 4 - 6 h (p. o. ) po ok. ½ h (i. m. , ok. 1 h s. c. )
 • • Dawka początkowa 60 mg 4 x/dobę Dawka maksymalna 600 -720")
Pirydostygmina (Mestinon) • • Dawka początkowa 60 mg 4 x/dobę Dawka maksymalna 600 -720 mg/dobę Zwykle dawki 240 - 480 mg/dobę Schemat dawkowania dostosowany indywidualnie

Leczenie immunosupresyjne • Kortykosteroidy • Azatiopryna • Inne leki immunosupresyjne – Cyklofosfamid – Cyklosporyna – Metotreksat – Mykofenolat mofetilu
 Wskazania: ü Przygotowanie do tymektomii ü")
Kortykosteroidy Lek immunosupresyjny pierwszego rzutu Preparat- prednizon (Encorton) Wskazania: ü Przygotowanie do tymektomii ü Brak zawodowalącej poprawy u chorego bez wskazań do tymektomii ü Brak poprawy po tymektomii

Schematy dawkowania prednizonu: Rozpoczynanie terapii: • dawka początkowa 1, 5 -2 mg/kg m. c. /dobę lub 6080 mg/dobę lub • osiągnięcie maksymalnej dawki stopniowo w ciągu 4 -6 tygodni Podanie wysokiej dawki kortykosteroidów może być przyczyną przejściowego pogorszenia stanu pacjenta

Schematy dawkowania prednizonu: • Utrzymanie wysokiej dawki do czasu osiągnięcia poprawy (zwykle 4 -6 tygodni) • Redukcja dawki: do 1 mg/kg m. c. / co 2 -gi dzień Dalsza powolna redukcja pod kontrolą stanu klinicznego: 5 -10 mg co 4 -8 tyg. (do dawki 30/0 mg) 5 mg co 4 -8 tyg. (do dawki 20/0 mg) 2, 5 mg co 4 -8 tyg.
 nie jest zalecany standardowo • wg niektórych autorów wskazany – jako początek")
Metylprednizolon (Solumedrol) nie jest zalecany standardowo • wg niektórych autorów wskazany – jako początek steroidoterapii – w przełomie miastenicznym • 0, 5 -2 g/dobę przez 3 -5 dni Podanie wysokiej dawki kortykosteroidów może być przyczyną przejściowego pogorszenia stanu pacjenta

Przewlekła steroidoterapia • profilaktyka osteoporozy: preparaty Ca+, Vit. D 3 lub bifosfoniany • • profilaktyka choroby wrzodowej monitorowanie ciśnienia tętnicznego, ocena glikemii, kontrola okulistyczna (jaskra, zaćma)

Przewlekła steroidoterapia • Leczenie prowadzone przez 2 -3 lata • Redukcja dawki prednizonu stopniowa, pod kontrolą stanu klinicznego Gwałtowne odstawienie prednizonu może być przyczyną przełomu miastenicznego
 Wskazania do terapii skojarzonej: • Brak poprawy po prednizonie (po ok. 3")
Azatiopryna (1) Wskazania do terapii skojarzonej: • Brak poprawy po prednizonie (po ok. 3 m-cach leczenia) • Konieczność odstawienia lub szybkiej redukcji prednizonu z powodu działań niepożądanych • Niepowodzenia w zmniejszaniu dawki prednizonu
 • • Dawka początkowa 2. 5 -3 mg/kg m. c. /dobę Dawka")
Azatiopryna (2) • • Dawka początkowa 2. 5 -3 mg/kg m. c. /dobę Dawka podtrzymująca 1. 5 -2. 5 mg/kg m. c. /dobę Efekt kliniczny najwcześniej po 3 -6 miesiącach Czas leczenia: co najmniej 2 -3 lata Pozwala na zmniejszenie dawki kortykosteroidów Okresowa kontrola morfologii i transaminaz Ryzyko choroby nowotworowej

Wskazania do tymektomii Grasiczak Miastenia uogólniona bez grasiczaka: ü brak dostatecznej poprawy po inhibitorach acetylocholinesterazy ü wiek 8 -55 lat ü pierwsze lata trwania choroby ü dodatnie p/ciała przeciwko ACh. R Brak dobrze udokumentowanych danych co do skuteczności leczenia Efekt możliwy do oceny po wielu latach !Operacja tylko w ośrodkach mających doświadczenie!
 Wniosek: „jako opcja w leczeniu uogólnionej")
Tymektomia w leczeniu MG bez grasiczaka (AAN 2000) Wniosek: „jako opcja w leczeniu uogólnionej MG zwiększająca szansę na remisję lub poprawę u pacjentów przed 60 r. ż. ” W leczeniu miastenii seropozytywnej?

Plazmafereza • • wymiana osocza w celu usunięcia krążących przeciwciał 3 -5 zabiegów wymiany osocza co drugi dzień wzór na objętość wymienianego osocza: V osocza (ml) = 80 ml x masa ciała (kg) x [100 – hematokryt (%)] płyn zastępczy - 5% roztwór albumin lub mrożone osocze

Przeciwwskazania • zaburzenia hemodynamiczne • zaburzenia krzepnięcia • uogólnione zakażenie

Iv. Ig • 0, 4 ml / kg masy ciała przez 5 dni = 2 g / kg masy ciała

Przypuszczalny mechanizm działania Iv. Ig w autoimmunologicznych chorobach neurologicznych • Immunomodulacja poprzez blokowanie receptora Fc komórek immunokompetentnych • Hamowanie patologicznej odpowiedzi immunologicznej poprzez • p-ciała antyidiotypowe rozpoznające idiotypy autoprzeciwciał i neutralizujące je • Zahamowanie wytwarzania przeciwciał przez limfocyty B poprzez wiązanie się przeciwciał antyidiotypowych z antygenami błony limfocytów B • Bezposredni wpływ na miejsce wiązania przeciwciał w neuronie, komórce mięśniowej i synapsie nerwowo-mięśniowej • Rozpuszczenie krążących kompleksów immunologicznych • Bezpośredni wpływ na komórki NK i limfocyty T supresorowe • Neutralizowanie antygenów wirusów i bakterii • Hamowanie układu dopełniacza poprzez wiązanie z Ig. G • Bezpośrednie wiązanie cytokin

Leczenie miastenii objawowe immunosupresyjne i immunomodulacyjne tymektomia

Leczenie miastenii objawowe inhibitory acetylocholinesterazy immunosupresyjne i immunomodulacyjne tymektomia

Leczenie miastenii objawowe inhibitory acetylocholinesterazy immunosupresyjne i immunomodulacyjne ükortykosteroidy üazatiopryna üinne leki immunosupresyjne: cyclofosfamid, cyklosporyna, mykofenolan mofetilu, metotreksat üimmunoglobulina podawana dożylnie üplazmafereza tymektomia

Miastenia wskazania do tymektomiii tak nie Nasilone objawy ? nie tak Kortykosteroidy, IVIg, PE poprawa tymektomia Brak poprawy farmakoterapia remisja farmakoterapia

Miastenia uogólniona bez wskazań do tymektomii brak poprawy po tymektomii Objawy umiarkowane Objawy łagodne Objawy ciężkie Kortykosteroidy Inhibitory acetylocholiesterazy IVIg, PE +/- azatiopryna poprawa? nie Inne terapie immunosupresyjne tak Zmniejszanie dawek leków

U 5 -10 % pacjentów z miastenią nie udaje się uzyskać zadowalającej poprawy, nawet po zastosowaniu pełnej immunoterapii !

Leczenie miastenii ocznej 1. Metody niefarmakologiczne leczenia opadania powiek i podwójnego widzenia 2. Leczenie farmakologiczne – objawowe: inhibitory acetylocholinesterazy – leki immunosupresyjne: prednizon, azatiopryna 3. Tymektomia – u większości chorych nie jest wskazana – konieczna u chorych z grasiczakiem

Leczenie miastenii ocznej • jeśli nie ma podejrzenia grasiczaka leczenie farmakologiczne • inhibitory ACh. E, ale często nieskuteczne, czasem nawet nasilenie lub ujawnienie dwojenia • dobra reakcja na leczenie immunosupresyjne (prednizon maks. 0, 5 mg/kg m. c. , azatiopryna)
 poprawa")
Miastenia oczna Inhibitory acetylocholinesterazy Brak poprawy Prednizon (02, -0, 5 mg/kg m. c/dobę) poprawa Tak Stopniowa redukcja dawki do minimalne dawki podtrzymującej nie Prednizon Metody niefarmakologiczne +azatiopryna

Iv. Ig można uznać za najbezpieczniejszą formę leczenia immunomodulującego u kobiet w ciąży Kwieciński i wsp. 1995 – Przełom miasteniczny w czasie ciąży Yamada i wsp. 2001 – Zespół Guillaina-Barré (Iv. Ig w 5 -tym dniu choroby)

MG Mu. SK-pozytywna: leczenie • dobra odpowiedź na PE, słabsza na Iv. Ig (Evoli 2003) • inhibitory acetylocholinesterazy, prednizon, azatiopryna mniej skuteczne niż w MG seropozytywnej • konieczne leczenie innymi lekami: cyklofosfamid, mykofenolan mofetilu (Evoli 2003, Sanders 2003, Zhou 2004)

LECZENIE PRZEŁOMU MIASTENICZNEGO • Hospitalizacja w OIT • Intubacja, wentylacja zastępcza – odstawienie inhibitorów ACh. E (drug holidays) – PE (3 -5 zabiegów wymiany osocza co drugi dzień) – Iv. Ig – immunosupresja (metylprednizolon lub prednizon) • Odłączenie od respiratora, ekstubacja Czas utrzymania rurki intubacyjnej?
 Iv. Ig=placebo (po")
Iv. Ig=PE (w leczeniu nagłego pogorszenia MG ocena po 4 tygodniach) Iv. Ig=placebo (po 6 tygodniach w łagodnej MG) Iv. Ig=metylprednisolon (w MG o średnim nasileniu objawów) PE=prednizon (po 4 tygodniach) konieczne dalsze kontrolowane badania oceniające skuteczność Iv. Ig oraz PE w leczeniu MG (Cochrane Review 2005)

wg Grob, 1999

leczenie miastenii ocznej • inhibitory acetylocholinesterazy często nieskuteczne, czasem nawet nasilenie lub ujawnienie dwojenia • dobra reakcja na leczenie immunosupresyjne (prednizon maks. 0, 5 mg/kg m. c. , azathiopryna) • jeśli nie ma podejrzenia grasiczaka wyłącznie leczenie farmakologiczne

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy tymektomia immunosupresja tymektomia

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy uogólniona łagodna MG tymektomia immunosupresja brak poprawy immunosupresja tymektomia

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy uogólniona łagodna MG tymektomia immunosupresja brak poprawy immunosupresja tymektomia

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy uogólniona łagodna MG tymektomia nasilone objawy opuszkowe/oddechowe immunosupresja brak poprawy immunosupresja tymektomia

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy uogólniona łagodna MG tymektomia brak poprawy immunosupresja nasilone objawy opuszkowe/oddechowe immunosupresja stabilizacja/poprawa tymektomia

leczenie miastenii uogólnionej inhibitory acetylocholinesterazy tymektomia immunosupresja tymektomia
 Wniosek: „jako opcja w leczeniu uogólnionej")
Tymektomia w leczeniu MG bez grasiczaka (AAN 2000) Wniosek: „jako opcja w leczeniu uogólnionej MG zwiększająca szansę na remisję lub poprawę u pacjentów przed 60 r. ż. ”
 do 1 mg/kg m. c. • azathiopryna")
immunosupresja/immunomodulacja • prednizon: (ambulatoryjnie w dawkach rosnących) do 1 mg/kg m. c. • azathiopryna ok. 2 mg/kg m. c. • cyklofosfamid • mycofenolan mofetilu (Cell. Cept) • metylprednizolon • IVIg • PE

Immunosupresja • leczenie prowadzone przez min. 1 -2 lata • redukcja dawki prednizonu stopniowa, pod kontrolą stanu klinicznego pacjenta • podanie wysokiej dawki, lub gwałtowne odstawienie prednizonu może być przyczyną przełomu miastenicznego
 IVIg=placebo (po 6 tygodniach")
IVIg=PE (w leczeniu nagłego pogorszenia MG ocena po 4 tygodniach) IVIg=placebo (po 6 tygodniach w łagodnej MG) IVIg=metylprednisolon (w MG o średnim nasileniu objawów) PE=prednisone (po 4 tygodniach) konieczne dalsze kontrolowane badania oceniające skuteczność IVIg oraz PE w leczeniu MG (Gajdos P, Chevret S, Toyka K, Cochrane Review 2004)

Kostera-Pruszczyk, 1997
- Slides: 116