Metabolisme Eritrosit dr Sri Lestari Sulistyo Rini MSc

Metabolisme Eritrosit dr Sri Lestari Sulistyo Rini, MSc
 constitutes 95% of the red blood cell’s dry")
HAEMOGLOBIN STRUCTURE Haemoglobin (MW 68, 000) constitutes 95% of the red blood cell’s dry weight. 65% of haemoglobin synthesis occurs during the nucleated stages of RBC maturation and 35% occurs during the reticulocyte stage.

Hemoglobin • A globular protein with an embedded heme group. • Synthesized in a series of steps: – Heme portion is sythesized in mitochondria and cytosol of immature RBC – Globin (protein) synthesized by ribosomes in the cytosol. – Hb production continues throughout early cell development (proerythroblast to reticulocyte) in the bone marrow.

STRUCTURE OF RBC. • Negative surface charge. • Bag of fluid with dissolved substances and hemoglobin • Membrane – – Outer glycoprotein coat – Lipid bilayer (PL 55%, Cholesterol 45%) • Inner protein molecules cytoskeleton – Spectrin, Actin, Ankyrin etc. • No sub cellular particles

Cytoskeleton of erythrocyte Erythrocyte cytoskeleton • provides shape, structure, permits stretch • 2 -D lattice of proteins links to membrane proteins: • spectrin (a, b) • actin • ankyrin • band 4. 1 • membrane proteins: • glycophorin • band 3 protein • Mature rbc does not synthesize new proteins • Gets lipids from circulating LDL

“Sickling” in Red Blood Cells

Physiological Characteristics and Functions of RBC Characteristics of RBC ③ Suspension stability: it cab be described by erythrocyte sedimentation rate (ESR) which is RBC descending distance per hour and suspension stability is inverse proportion to ESR. Normal value of ESR: male, 0~15 mm/h; female, 0~20 mm/h. ESR and clinic: some diseases bring about rouleaux formation (mainly involved in plasma component, e. g. globulin, fibrinogen, cholesterol) and speed up ESR.
 and")
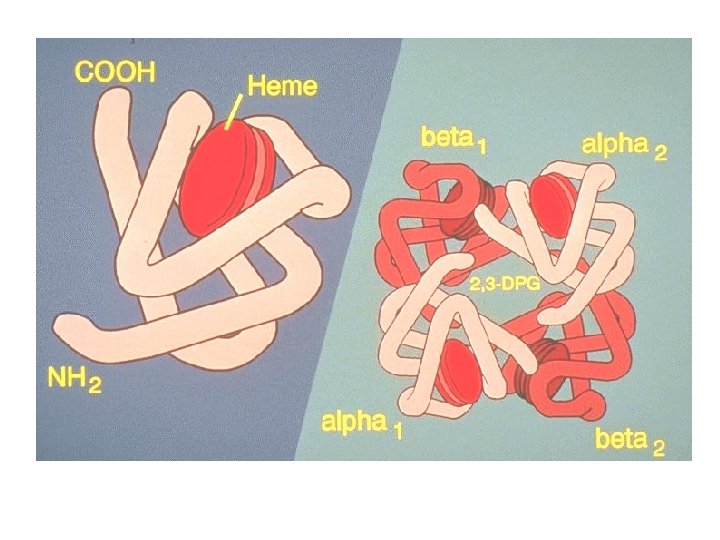
Normal Hb consists of globin (a tetramer of two pairs of polypeptide chains) and four haem groups. Each haem group contains a protoporphyrin ring plus ferrous iron (Fe++). The mitochondria are the main site of protoporphyrin synthesis. Iron is supplied from circulating transferrin and globin chains are synthesised on ribosomes.

Finally, ferrous iron is inserted to form haem by haem synthetase. The ferrous iron atom in each haem molecule is attached to the proximal histidine residue of a globin chain, but not to the distal histidine residue. Amino acids lying in the loop between proximal and distal histidine residues form the haem pocket, essential for the O 2 carrying capacity.

Six Hb variants are normally formed. Embryonic haemoglobins include Gower 1, Gower 2 and Hb Portland. Hb. F is the predominant haemoglobin of fetal life (65 -95%). Adults have only trace amounts of Hb. F (<1%). Hb. A (>95%) and Hb. A 2 (2. 5 -3. 5%) are the main adult haemoglobins.
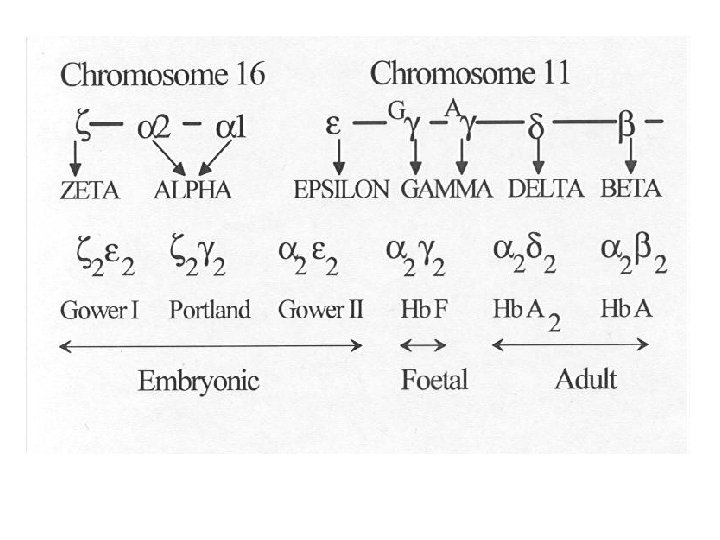

Alpha chain synthesis is directed by two α genes, α 1 and α 2, on chromosome 16. Beta and delta chains result from single genes on chromosome 11. The gamma chain is directed by two genes, G and A, on chromosome 11. Alpha chains have 141 amino acids and nonalpha chains have 146; the exact sequence of the amino acids has been determined.

HAEM consists of four pyrrole rings with a central iron atom linked to the four nitrogen atoms. The iron atom has two further binding sites, one of which is bound to a globin histidine residue and the other binds reversibly to oxygen.

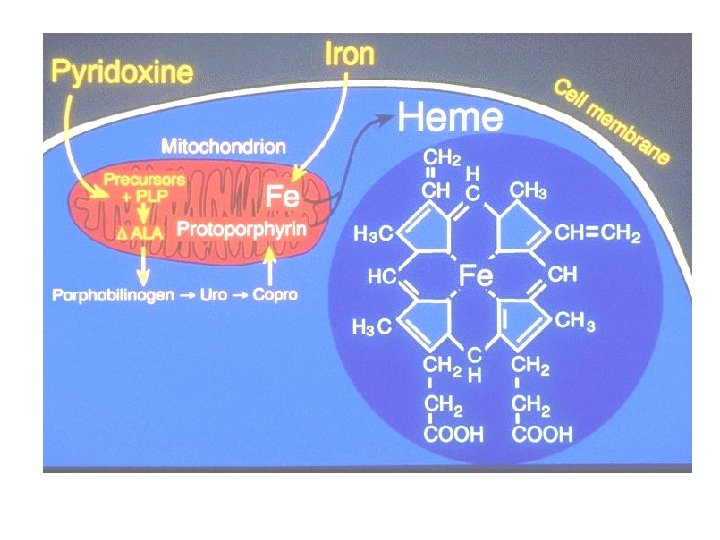
Haem is synthesised mainly in mitochondria of erythroblasts, some steps occur in the cytoplasm. The initial and rate-limiting step is the fusion of succinyl-Co A with glycine mediated by ALA synthetase to form δ-aminolaevulinic acid (ALA). This occurs in the mitochondrion and depends on the presence of vitamin B 6 (pyridoxal phosphate). The reaction is stimulated by erythropoietin and inhibited by haem.

The next step occurs in the cytoplasm. Two molecules of ALA fuse to form porphobilinogen. A double enzyme step forms uroporphyrinogen that is decarboxylated to coproporphyrinogen. At this point, the pathway re-enters the mitochondrium where protoporphyrin is formed.
 beta (β) gamma (γ)")
Globin chains found in adult haemoglobins are designated alpha (α) beta (β) gamma (γ) delta (δ). Hb A has two alpha and two beta chains (α 2β 2), Hb F has two alpha and two gamma chains (α 2γ 2) Hb A 2 has two alpha and two delta chains (α 2δ 2). The alpha chain is thus common to all three types of adult Hb.

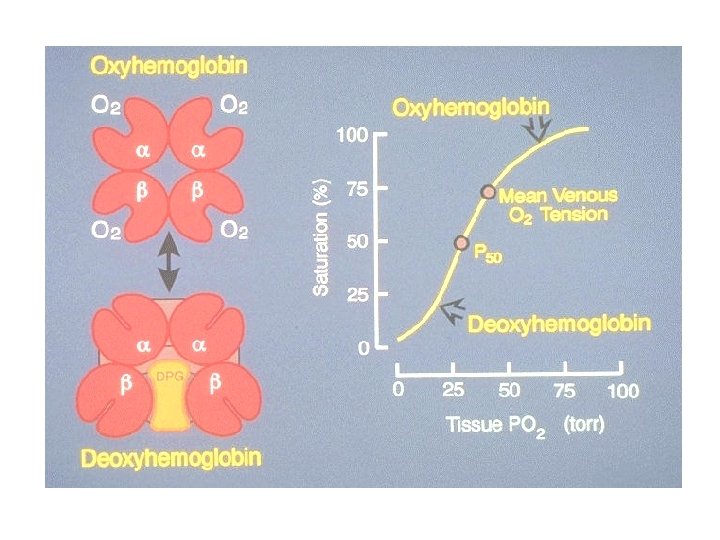
HAEMOGLOBIN FUNCTION Red cells carry oxygen from the lungs to the tissues and return in venous blood with carbon dioxide. As the haemoglobin molecule loads and unloads O 2, the individual globin chains in the haemoglobin molecule move in relation to each other. When O 2 is unloaded, the β chains are pulled apart, permitting entry of the metabolite 2, 3 diphosphoglycerate (2, 3 -DPG) resulting in a lower affinity for O 2.

During oxygenation the two β chains move together to give a species more avid for O 2. Hence as the initial oxygen is taken up by Hb it increases its affinity for oxygen to bind to the remaining haem groups in the molecule. The relationship between oxygen concentration in the blood (the partial pressure of oxygen) and the proportion of oxygen bound to haemoglobin (the percentage oxygen saturation) forms a sigmoid curve called the Hb oxygen-dissociation curve.

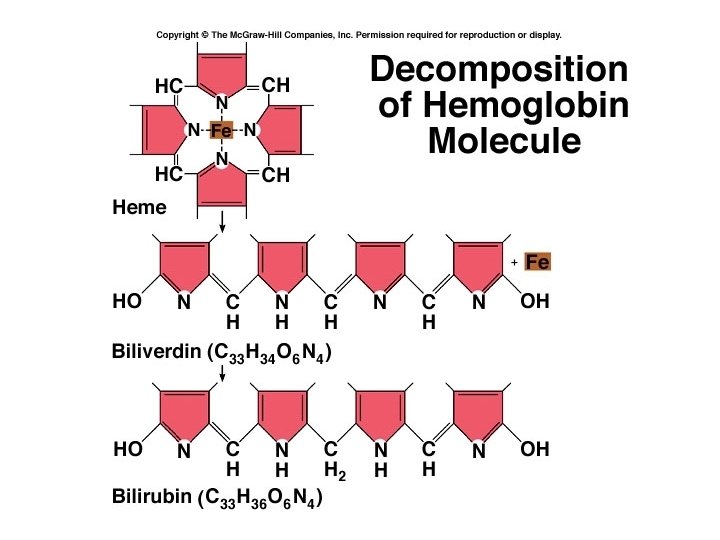
RBC life span and circulation • Replaced at a rate of approximately 3 million new blood cells entering the circulation per second. • Replaced before they hemolyze • Components of hemoglobin individually recycled – Heme stripped of iron and converted to biliverdin, then bilirubin • Iron is recycled by being stored in phagocytes, or transported throughout the blood stream bound to transferrin

Life and breakage of RBC • Life-span: 120 days, about 4 months, each RBC circulates 27 km averagely in vessels, short life-span for aged RBC • Breakage: places are liver, spleen and lymphatic node, and after breakage, Hb released from RBC immediately combine with plasma α 2 -globulin (Hb touched protein) which is taken in by liver for iron reuse. • Hb, very toxic if it get into blood, normally, it can be metabolized into bile pigment in liver. • Clinic relation.

RBC breakdown • Healthy RBC’s live about 120 days; we break down about 174 million per minute • RBC’s are removed from circulation by the liver and spleen • Broken down into heme and globin portions • Globin is broken down into amino acids • Iron is removed from heme and stored or recycled • Heme is broken down into biliverdin and then into bilirubin

Life Cycle of Red Blood Cells Figure 18. 7
 Phagocytosis & Lysis Hemoglobin")
Extravascular Pathway for RBC Destruction (Liver, Bone marrow, & Spleen) Phagocytosis & Lysis Hemoglobin Globin Heme Amino acids Fe 2+ Amino acid pool Bilirubin Excreted

Degradation of hemoglobin • Heme is degraded to bilirubin: • Bilirubin is congugated to glucuronate (more soluble), excreted • Rbc only live ~120 days • Globin is degraded to amino acids Figs. 7, 8

DEGRADATION OF HEME TO BILIRUBIN 75% is derived from RBCs P 450 cytochrome In normal adults this results in a daily load of 250 -300 mg of bilirubin Normal plasma concentrations are less then 1 mg/d. L Hydrophobic – transported by albumin to the liver for further metabolism prior to its excretion “unconjugated” bilirubin

Heme synthesis • Heme synthesis begins with d-ALA: • Decarboxylation by d-ALA synthase • PLP is pyridoxal phosphate • Dehydratase joins 2 d-ALA • 4 pyrroles form porphyrinogen Fig. 4

Figure 19. 5

Iron metabolism • Iron metabolism: • Transferrin carries Fe 3+ to cells; stored as ferritin • Transferrin taken up by R-mediated endocytosis • Hemosiderin stores excess Fig. 6 RE = reticuloendothelial system
")
• Heme synthesis: • • Glycine, succinyl Co. A form d-Aminolevulinic acid (d-ALA) Each heme needs 8 of each • • Final step is Fe 2+ Heme regulates: inhibit 1 st enzyme repress synthesis • • Porphyria diseases from defective enzymes • • • intermediates accumulate photosensitive, toxic products Fig. 3

NORMAL BILIRUBIN METABOLISM Uptake of bilirubin by the liver is mediated by a carrier protein (receptor) Uptake may be competitively inhibited by other organic anions On the smooth ER, bilirubin is conjugated with glucoronic acid, xylose, or ribose Glucoronic acid is the major conjugate - catalyzed by UDP glucuronyl tranferase “Conjugated” bilirubin is water soluble and is secreted by the hepatocytes into the biliary canaliculi Converted to stercobilinogen (urobilinogen) (colorless) by bacteria in the gut Oxidized to stercobilin which is colored Excreted in feces Some stercobilin may be re-adsorbed by the gut and re-excreted by either the liver or kidney
 occurs when there is")
HYPERBILIRUBINEMIA Increased plasma concentrations of bilirubin (> 3 mg/d. L) occurs when there is an imbalance between its production and excretion Recognized clinically as jaundice

|haematinics: the normal iron cycle 2. 4 Partners in Global Health Education Iron deficiency can be identified best by assessing the appearances of the red cells on a blood film. Iron indices in a blood sample are helpful to confirm a lack of iron. In order to interpret these indices, it is vital to understand how the body handles iron …. . Iron is a key constituent of haemoglobin (60 -70% of total body iron is stored here) and it’s availability is essential for erythropoiesis. In iron deficiency, there are more divisions of red cells during erythropoiesis than normal. As a result the red cells are smaller (microcytic) and have a reduced haemoglobin content (hypochromic). Soluble transferrin receptors, s. Tf. R are on the red cell surface. These can be measured and are increased in iron deficiency. Red blood cells In iron deficient states, bone marrow iron is reduced. Erythroid bone marrow (normoblasts) Some iron binds to apoferritin to form ferritin, a storage compound. Liver 2. Iron is then attached to a protein, transferrin in the serum (plasma), where it is transported to the bone marrow for haemoglobin synthesis. Serum transferrin Fe Duodenum Reticuloendothelial system; Spleen & macrophages 3. Dying red cells are recycled by macrophages in the spleen and iron is recycled into the plasma for further use. 1. Iron is absorbed from the small intestine in the ferrous state (Fe 2+; approx. 1 mg/day). START

|haematinics: vitamin B 12 2. 4 Partners in Global Health Education There a number of key steps in the absorption of Vitamin B 12. The two key locations are the stomach and the terminal ilium. Dietary vitamin B 12 binds with intrinsic factor (IF) in the stomach, a transport protein produced by gastric parietal cells. The B 12 -IF complex then travels through the small intestine and is absorbed by special receptors in the distal ileum. This pathway is important when considering possible causes of Vitamin B 12 deficiency. Oesophagus Causes of vitamin B 12 deficiency 1. Pernicious anaemia Stomach IF Intrinsic factor 2. Inadequate intake 3. Poor absorption Distal ileum Site of B 12 absorption Click here to return Vitamin B 12 ingested Vitamin B 12 deficiency can take up to two years to develop as the body has sufficient stores for this period. Pernicious anaemia: the leading cause of B 12 deficiency. Ig. G autoantibodies target gastric parietal cells and its product IF causing an atrophic gastritis. This results in reduced secretion of intrinsic factor and therefore reduced B 12 -IF complex for absorption in the distal ileum.

1 Low O 2 levels in blood stimulate kidneys to produce erythropoietin. 2 Erythropoietin levels rise in blood. 3 Erythropoietin and necessary raw materials in blood promote erythropoiesis in red bone marrow. 4 New erythrocytes enter bloodstream; function about 120 days. Figure 17. 7, step 4

5 Aged and damaged red blood cells are engulfed by macrophages of liver, spleen, and bone marrow; the Bilirubin hemoglobin is broken down. Hemoglobin Heme Globin Amino Iron stored acids as ferritin, hemosiderin Iron is bound to transferrin and released to blood from liver as needed for erythropoiesis. Bilirubin is picked up from blood by liver, secreted into intestine in bile, metabolized to stercobilin by bacteria, and excreted in feces. Circulation Food nutrients, including amino acids, Fe, B 12, and folic acid, are absorbed from intestine and enter blood. 6 Raw materials are made available in blood for erythrocyte synthesis. Figure 17. 7, step 6

1 Low O levels in blood stimulate 2 kidneys to produce erythropoietin. 2 Erythropoietin levels rise in blood. 3 Erythropoietin and necessary raw materials in blood promote erythropoiesis in red bone marrow. 5 Aged and damaged red blood cells are engulfed by macrophages of liver, spleen, and bone marrow; the hemoglobin Hemoglobin is broken down. Heme Bilirubin 4 New erythrocytes enter bloodstream; function about 120 days. Globin Amino Iron stored acids as ferritin, hemosiderin Iron is bound to transferrin and released to blood from liver as needed for erythropoiesis. Bilirubin is picked up from blood by liver, secreted into intestine in bile, metabolized to stercobilin by bacteria, and excreted in feces. Circulation Food nutrients, including amino acids, Fe, B 12, and folic acid, are absorbed from intestine and enter blood. 6 Raw materials are made available in blood for erythrocyte synthesis. Figure 17. 7

Key point: Oxidant stress! CLICK HERE H 2 O O- |red cell metabolism 2. 5 Partners in Global Health Education Contents page Embden-Meyerhof glycolytic pathway 2. 1. The erythrocyte: an overview. 2. 2. Erythropoiesis 2. 3. The red cell structure 2. 3. 1. Cell membrane 2. 3. 2. DNA synthesis 2. 4. Red cell metabolism 2 GSH GSSG Hexose shunt. Glucose NAPD NADPH+H+ Glucose- 6 -PG Glucose-6 -phosphate dehydrogenase ADP monophosphate Red cells require a mechanism to detoxify the waste products (accumulated oxidised substrates) of the cell. This shunt provides this solution. It also provides 10% of glycolysis. However this metabolic pathway is also susceptible to pathology. ATP Key point! This is a sequence of biochemical reactions in which glucose is metabolised to lactate with the generation of 2 ATP molecules (providing energy for the cell). Fructose-6 -P Ribulose 5 -P ADP Pyruvate kinase ATP Hexose monophosphate shunt The glycolytic pathway With no cell organelles and no mitochondria the fully developed erythrocyte relies on this aerobic pathway to gain energy (ATP) for the cell. Lactate Pyruvate kinase deficiency: In rare circumstances there are defects within the critical glycolytic enzymes. 95% of these defects are associated with pyruvate kinase, a key enzyme within this pathway. The result is insufficient ATP production for cell life and therefore premature death (haemolysis). Glucose-6 -phosphate dehydrogenase (G 6 PD) deficiency is an X-linked disorder that is relatively common. The G 6 PD enzyme is a rate-limiting step within this pathway. If deficient, haemolysis occurs when the cell is placed under oxidative stress (e. g. by oxidative drugs, fava beans, infections) creating a potentially severe anaemia. Click OXIDATIVE STRESS for more info.

ENERGY METABOLISM • Less energy required – Na + - K + pump – Iron in Fe ++ form • Utilize Glucose by GLUT 1 • Anaerobic respiration – Glycolysis – Embden Meyerhof pathway • Pentose phosphate pathway. – Hexose monophosphate shunt

Erythrocyte metabolism • Erythrocyte metabolism: • • ATP for Na+/K+, Ca 2+ HMP shunt makes NADPH G 6 PD is 1 st enzyme Lifetime rbc by G 6 PD activity 2, 3 -BPG modulates O 2 binding Need Fe 2+ Hb bind O 2; If ROS made Fe 3+, NADH can reduce Only glycolysis

2, 3 -Bisphoglycerate Pathway

Glycolysis or Embden-Meyerhof Pathway • Generates 90 - 95% of energy needed by RBC’s • Glucose is metabolized and generates two molecules of ATP (energy). • Functions in the maintenance of RBC shape, flexibility and the cation pumps

Hexose monophosphate shunt Metabolizes 5 -10% of glucose. NADPH is end product Protects the RBC from oxidative injury. Most common defect is deficiency of the enzyme glucose-6 -phosphate dehydrogenase (G-6 PD). • If the pathway is deficient, intracellular oxidants can’t be neutralized and globin denatures then precipitates. The precipitates are referred to as Heinz bodies • •
 state. • In the")
Methemoglobin Reductase pathway • Maintains iron in the ferrous (Fe++) state. • In the absence of the enzyme (methemoglobin reductase), methemoglobin accumulates and it cannot carry oxygen.

Rapoport –Leubering Shunt • Allows the RBC to regulate oxygen transport during conditions of hypoxia or acid-base imbalance. • Permits the accumulation of 2, 3 -DPG which is essential for maintaining normal oxygen tension, regulating hemoglobin affinity
 Hemoglobin Purposes: 1. Scavenge iron 2. Prevent major iron losses")
Handling of Free (Intravascular) Hemoglobin Purposes: 1. Scavenge iron 2. Prevent major iron losses 3. Complex free heme (very toxic) • Haptoglobin: hemoglobin-haptoglobin complex is readily metabolized in the liver and spleen forming an iron-globin complex and bilirubin. Prevents loss of iron in urine. • Hemopexin: binds free heme. The heme-hemopexin complex is taken up by the liver and the iron is stored bound to ferritin. • Methemalbumin: complex of oxidized heme and albumin.

When the erythrocyte is destroyed within the vascular system, hemoglobin is released directly into the blood. Normally, the free hemoglobin quickly complexes with haptoglobin, and the complex is degraded in the liver. In severe hemolytic states, haptoglobin can become depleted, and free hemoglobin dimers are filtered by the kidney. Additionally, with haptoglobin depletion, some hemoglobin is quickly oxidized to methemoglobin and bound to either hemopexin or albumin for eventual degradation in the liver.
- Slides: 50