Metabolism of Sulfur containing amino acids Dr Ketki

Metabolism of Sulfur containing amino acids Dr. Ketki K Assistant Professor Dept of Biochemistry HIMS Varanasi

Sulfur containing amino acids Ø The sulfur – containing amino acids: Methionine, cysteine & cystine Ø Methionine is glucogenic & essential amino acid Ø Serves as a precursor for the synthesis of cysteine & cystine which are non-essential Ø Cysteine & cystine are interconvertible Ø Cystine is found exclusively in protein

Ø Methionine: required for the initiation of protein biosynthesis Ø The sulfur - containing amino acids: almost an exclusive dietary source of sulfur to the body

Metabolism of Methionine It may be divided into three parts 1. Activation of methionine & transmethylation reaction 2. Conversion of methionine to cysteine & cystine 3. Degradation of cysteine & its conversion to specialized products

Synthesis of SAM

Methionine metabolism Methionine adenosyltransferase Methionine 2 Pi + Pi ATP THF SAM Methyl transferase N 5 -Methyl THF Acceptor CH 3 Methylated product Adenosylhomocysteinase Homocysteine Adenosine S-adenosylhomocysteine H 2 O
 or")
q. Activation of methionine Ø Methionine has to be activated to Sadenosylmethionine (SAM) or active methionine [to donate the methyl group] Ø The synthesis of SAM occurs by the transfer of adenosyl group from ATP to sulfur atom of methionine
 Ø 3 isoenzymes for MAT")
Ø This reaction is catalysed by methionine Sadenosyltransferase (MAT) Ø 3 isoenzymes for MAT Ø Three high energy phosphates (3 ATP) are consumed in the formation of SAM Ø SAM is the main source of methyl groups in body

q Methyl transfer: Ø SAM transfers the methyl group to an acceptor & gets itself converted to S-adenosyl-homocysteine (SAH) → enzyme: methyltransferase q Homocysteine: Ø S-Adenosylhomocysteine (SAH) is hydrolysed (adenosyl group is removed) to homocysteine & adenosine → enzyme: adenosine homocysteinase
: Ø Homocysteine can be remethylated to methionine by N")
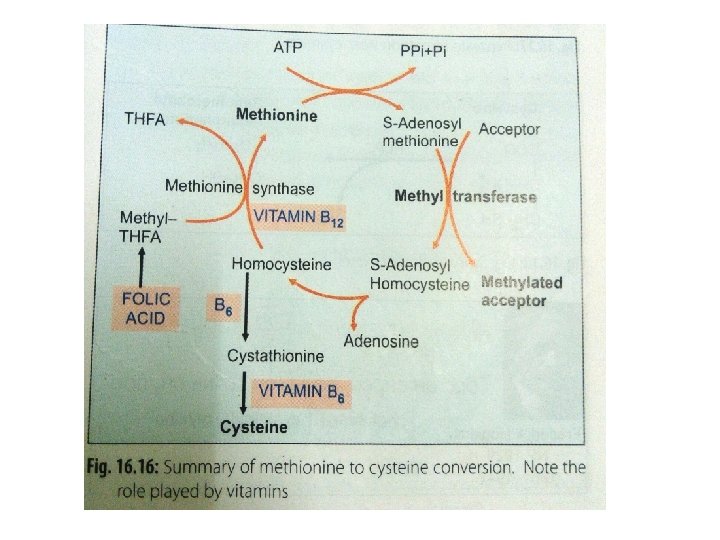
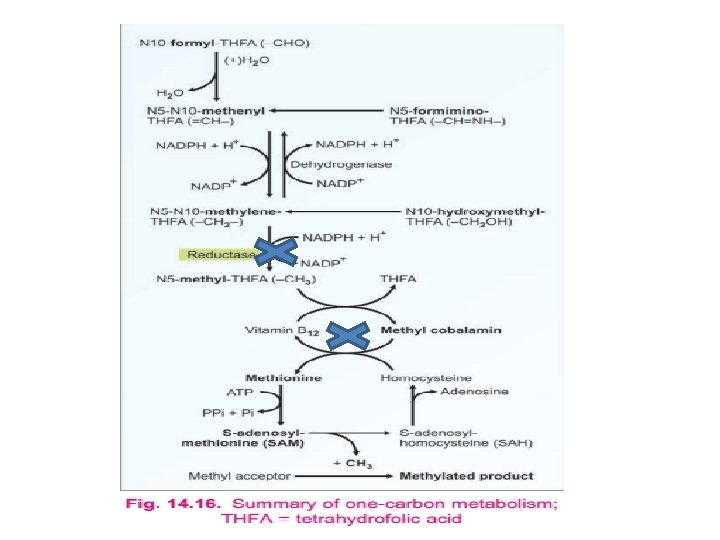
q Methionine synthesis (from homocysteine): Ø Homocysteine can be remethylated to methionine by N 5 -methyl tetrahydrofolate Ø This methyl group is donated from one-carbon pool, with the help of vitamin B 12 Ø In this manner, methionine can be regenerated for reuse

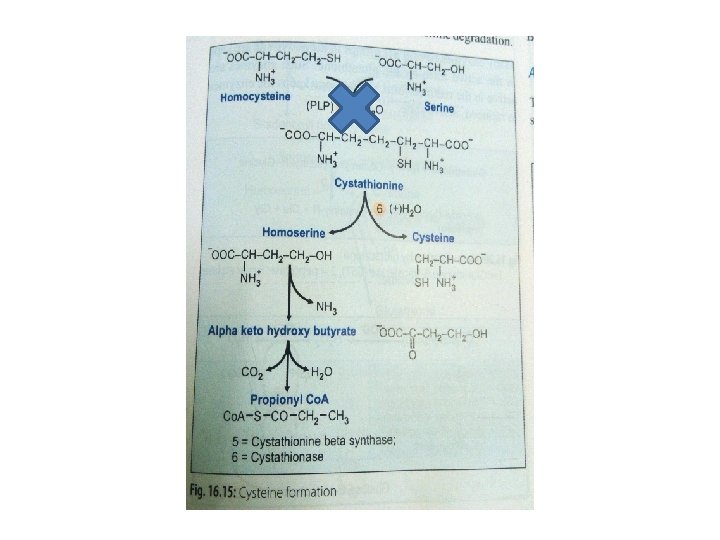
q Homocysteine degradation: Ø Homocysteine condenses with serine to form cystathionine Ø This is catalysed by PLP dependent cystathionine-βsynthase Ø Absence of this enzyme leads to homocystinuria

q. Cysteine synthesis: Hydrolysis of cystathionine to form homoserine & cysteine → enzyme: cystathionase Net result: SH group is transferred from methionine to serine to form cysteine It is c/a trans-sulfuration reaction

q. Final oxidation Homoserine is deaminated & then decarboxylated to form propionyl Co. A Which enters into TCA cycle as succinyl Co. A Which is converted to glucose

 from active methionine/SAM to an")
Transmethylation Ø The transfer of methyl group (-CH 3) from active methionine/SAM to an acceptor is known as transmethylation Ø SAM acts as donor of methyl group to the acceptor , so that acceptor is converted to methyl acceptor Ø The enzymes involved in the transfer of methyl group : collectively k/a methyl-transferases

Ø Origin of methyl group: derived from one carbon pool Ø Methyl THFA can transfer methyl group to homocysteine Ø Vit B 12 acts as co-enzyme Ø Explain folate trap

Transmethylation reactions Methyl group acceptor Methylated product Guanidinoacetate Creatine Norepinephrine Epinephrine Metanephrine Ethanolamine Choline Nicotinamide N-Methylnicotinamide Acetyl serotonin Melatonin Phosphatidylethanolamine Phosphatidylcholine Serine Choline Carnosine Anserine t-RNA bases Methylated t-RNA bases Lysine Methyl lysine

• "As soon as the fear approaches near, attack and destroy it. " Chanakya quotes (Indian politician, strategist and writer, 350 BC-275 BC)

Cysteine Metabolism
 2) 3) 4) 5) Introduction Synthesis of cysteine Degradation of cysteine Metabolic")
Content 1) 2) 3) 4) 5) Introduction Synthesis of cysteine Degradation of cysteine Metabolic function of cysteine Disorders a/w cysteine metabolism
(C) Ø Non-essential Ø Glucogenic amino acid Ø Cysteine : present in large")
Cysteine (Cys)(C) Ø Non-essential Ø Glucogenic amino acid Ø Cysteine : present in large quantity in keratin of hair & nails Ø Formation of cysteine is by using the carbon skeleton contributed by serine & sulfur originating from methionine

Synthesis of Cysteine
 Trans-amination 2) May be removed as H 2 S 3)")
Degradation of Cysteine 1) Trans-amination 2) May be removed as H 2 S 3) Decarboxylation

Pyruvate formation from cysteine

Decarboxylation of cysteine
 Amino acid transport 2) Co-enzyme role")
Metabolic Functions of Cysteine Formation of Glutathione 1) Amino acid transport 2) Co-enzyme role 3) RBC membrane integrity 4) Prevents met-hemoglobinemia 5) Conjugation for detoxification 6) Activation of enzymes 7) Formation of Taurine 8) Keeping correct structure of proteins

Glutathione

Role of glutathione in Amino acid transport
→ GS-SG (Oxidized) + H 2")
Role of glutathione as Co-enzyme 2 GSH ( Reduced)→ GS-SG (Oxidized) + H 2 Hydrogen released is used for reducing other substrates 1) Maleylacetoacetate → Fumarylacetoacetate 2) Cysteic acid → Taurine 3) (Iodine) I 2 + 2 GSH → 2 HI + GS-SG

Role of glutathione in maintaining RBC membrane integrity Free radical scavenging
 + 2 GSH → 2")
Role of glutathione in preventing methemoglobinemia 2 Met-Hb-(Fe 3+) + 2 GSH → 2 Hb –(Fe 2+) + 2 H + + GS-SG Glutathione is required to convert met. Hb(ferric state) to normal Hb (ferrous state)

Role of glutathione in conjugation for detoxification Helps to detoxify several compounds by transferring the cysteinyl group, Eg: 1) Organophosphorus compounds 2) Halogenated compounds 3) Heavy metals 4) Drug metabolism
 GST 2) Peptidase 3) Acetylase")
1) GST 2) Peptidase 3) Acetylase

• Role of glutathione in keeping the enzymes in reduced , active state

Formation of taurine Ø Cysteine to cystic acid and then taurine Ø Taurine is used for conjugation of bile acids Ø Taurine + Cholyl Co. A → Tauro cholate + Co. ASH Ø Taurine: Inhibitory Neurotransmitter in CNS

Formation of taurine from cysteine

Role of glutathione in Keeping correct structure of proteins Cysteine residues in polypeptide chains form disulfide bridges to make active proteins, e. g. insulin & immunoglobulins
 2) 3) 4) Cystinuria Cystinosis")
Inborn errors of sulfur containing amino acid metabolism 1) 2) 3) 4) Cystinuria Cystinosis Homocystinuria Cystathioninuria Acquired Hyperhomocysteinemia
: • The most common")
Inborn errors of sulfur amino acid metabolism q Cystinuria (cystine-lysinuria): • The most common inherited disease/AR • It is characterized by increased excretion of cystine • Elevation in the urinary output of ornithine, arginine & lysine

Ø A specific carrier system exists in kidney tubules for the reabsorption of amino acids, namely cystine, ornithine, arginine & lysine (COAL to recall) Ø In cystinuria, this carrier system becomes defective leading to the excretion of all these four amino acids in urine (cause)

Ø Cystine is relatively insoluble & increased concentrations leads to precipitation & formation of cystine stones in kidney & urinary tract (c/f) Ø Cystinuria is usually identified in the laboratory by cyanide nitroprusside test Ø The treatment includes restricted ingestion of dietary cysteine & high intake of fluids
 Ø Cystine crystals are deposited in many tissues & organs")
Cystinosis (cystine storage disease) Ø Cystine crystals are deposited in many tissues & organs of reticulo-endothelial system throughout the body Ø These include spleen, lymph nodes, liver, kidney, bone marrow etc. Ø A defect in the Iysosomal function Ø Cystine accumulates in the lysosomes of various tissues

Ø In cystinosis, renal function is impaired Ø It is characterized by generalized amino aciduria Ø The affected patients die within 10 years, mostly due to renal failure

Homocystinurias Ø Homocystinurias : A group of metabolic disorders characterized by the accumulation & increased urinary excretion of homocysteine & S-adenosyl methionine, decreased cysteine in urine Ø Plasma concentration of methionine is increased

Homocystinuria type I ü Enzyme defect: Cystathionine beta synthase Ø Accumulation of homocysteine & methionine, excreted in urine Ø Plasma cysteine reduced ü Two forms of type I homocystinurias One of them can be corrected with vitamin B 6 supplementation (B 6 responsive) while the other does not respond to B 6.

Homocystinuria ll: • N 5 - N 10 - Methylene THF reductase deficiency Homocystinuria lll: • N 5 Methyl THF homocysteine methyltransferase deficiency • This is mostly due to impairment in the synthesis of methylcobalamin Homocystinurla l. V: • N 5 - Methyl THF homocysteine methyl transferase deficiency • This is primarily due to a defect in the intestinal absorption of vitamin B 12
 Mental retardation 2) Charley chaplin gait/ Skeletal deformity 3) Ectopia lentis, myopia,")
C/F: 1) Mental retardation 2) Charley chaplin gait/ Skeletal deformity 3) Ectopia lentis, myopia, glaucoma 4) Thrombosis
 Aminoaciduria: Homocysteine in urine 2) Increased methionine & homocysteine(normal level: 5 -15")
Investigation: 1) Aminoaciduria: Homocysteine in urine 2) Increased methionine & homocysteine(normal level: 5 -15 µmol/L) in blood 3) Cyanide-nitroprusside test: positive in urine , it indicates urine homocysteine > 300 mg/24 h

Treatment: Ø The treatment includes consumption of diet low in methionine & high in cysteine Ø PLP given in large doses (500 mg) : corrects the defect

• The patients of homocystinuria have high levels of homocysteine & usually die of myocardial infarction, stroke. • Homocysteine & heart attack:

Homocysteine & heart attack Homocysteine interacts with lysyl residue of collagen ↓ Interfere with collagen cross linking & also defective protein fibrillin ↓ forms homocysteine thiolactone ↓ which binds with LDL particles ↓ Particles aggregate & endocytosed by macrophages ↓ increased tendency for atherogenesis

• Providing adequate PLP, Folic acid, Vitamin B 12 keep homocysteine in blood at normal level • Maternal hyperhomocysteinemia: Increase chance of neural tube defect in fetus, so high doses of folic acid advised during pregnancy

Cystathioninuria Autosomal recessive condition Enzyme defect: cystathionase deficiency C/F: Mentalretardation, anemia, thrombocytopenia Investigation: Cystathionine in blood & urine, Treatment: cysteine rich diet, diet low in methionine, PLP in large doses(200 -400 mg)
 Nutritional: B 12, folic acid, PLP deficiency 2) Metabolic: hypothyroidism,")
Acquired Hyperhomocysteinemia Causes 1) Nutritional: B 12, folic acid, PLP deficiency 2) Metabolic: hypothyroidism, chronic renal disease 3) Drug induced: Folate antagonist, B 12 antagonist, Pyridoxine antagonist • Oasthouse syndrome is due to malabsorption of methionine. Such children excrete methionine, aromatic & branched chain amino acids in urine

Thank You
- Slides: 58