Metabolism of some selected amino acids Histidine Metabolism

Metabolism of some selected amino acids

Histidine Metabolism: Histamine Formation Histidine decarboxylase Histidine CO 2 Histamine: • Synthesized in and released by mast cells • Mediator of allergic response: vasodilation, bronchoconstriction (H 1 receptors) • H 1 blockers: Diphenhydramine (Benadryl) Loratidine (Claritin) • Stimulates secretion of gastric acid (H 2 receptors) • H 2 blockers: Cimetidine (Tagamet); ranitidine (Zantac)
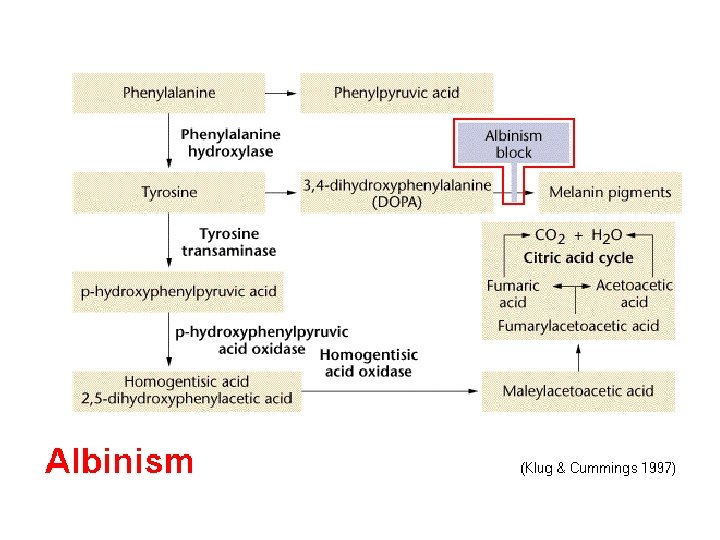
 Phenylketonuria (PKU) Disease (Phenylalanine")
آﻼﻧیﻦ ﻭ ﺗیﺮﻭﺯیﻦ ﻣﺘﺎﺑﻮﻟیﺴﻢ ﻓﻨیﻞ Phenyl ketones Phenylalanine (Essential) Phenylketonuria (PKU) Disease (Phenylalanine hydroxylase) ﺳﻨﺘﺰ ﺍﻧﻮﺍﻉ پﺮﻭﺗﺌیﻨﻬﺎ ﺑﺎﻓﺘی + ﺗیﺮﻭﺋیﺪ DOPA Tyrosine Tyrosinase Melanine ( ﺍﻟکﺎپﺘﻮﻥ Homogentisate Alkaptonuria × Norepinephrine Homogentisate dioxygenase Fumarate + acetoacetate ATP T 3& T 4 NADPH + H+ Albinism Epinephrine (Adrenaline) ﺳﻨﺘﺰ ﺍﻧﻮﺍﻉ پﺮﻭﺗﺌیﻨﻬﺎ ﺑﺎﻓﺘی ﺳیکﻞ کﺮﺑﺲ

Biosynthesis of Tyrosine from Phenylalanine hydroxylase is a mixed-function oxygenase: one atom of oxygen is incorporated into water and the other into the hydroxyl of tyrosine. The reductant is the tetrahydrofolate-related cofactor tetrahydrobiopterin, which is maintained in the reduced state by the NADH-dependent enzyme dihydropteridine reductase

Phenylketonuria Hyperphenylalaninemia - complete deficiency of phenylalanine hydroxylase (plasma level of Phe raises from normal 0. 5 to 2 mg/d. L to more than 20 mg/d. L). The mental retardation is caused by the accumulation of phenylalanine, which becomes a major donor of amino groups in aminotransferase activity and depletes neural tissue of α-ketoglutarate. Absence of α-ketoglutarate in the brain shuts down the TCA cycle and the associated production of aerobic energy, which is essential to normal brain development. Newborns are routinelly tested for blood concentration of Phe. The diet with low-phenylalanine diet.

Alternative pathways of phenylalanine catabolism in phenylketonuria

Homogentisic Acid Formation Transamination Tyrosine p-Hydroxyphenylpyruvate O 2 Deficient in alkaptonuria Cleavage of aromatic ring Homogentisate dioxygenase Fumarate + acetoacetate p-Hydroxyphenylpyruvate dioxygenase (ascorbate-dep. ) CO 2 Homogentisate

Alkaptonuria • First defect to which inborn error of metabolism applied – Sir Archibald Garrod in early 1900’s • Homogentisate appears in urine • Deposited in cartilage and elsewhere polymerization (black) • Deficiency of homogentisate dioxygenase • Urine turns dark on standing • Oxidation of homogentisic acid • Asymptomatic in childhood • Tendency toward arthritis in adulthood
 DOPA decarboxylase Epinephrine (Adrenaline)")
Catecholamine Biosynthesis Catechol Tyr hydroxylase O 2 Tyrosine Dihydroxyphenylalanine (DOPA) DOPA decarboxylase Epinephrine (Adrenaline) Methyl transferase S-Adenosylhomocysteine SAM DOPA, dopamine, norepinephrine, and epinephrine are all neurotransmitters CO 2 Dopamine hydroxylase Dopamine Norepinephrine 10

L-DOPA in Parkinsonism Blood L-DOPA Brain L-DOPA Dopamine Blocks Parkinsonism associated with dopamine in brain through loss of neurons in basal ganglia. Carbidopa + L-DOPA Carbidopa Dopamine Blood Brain Barrier 11
 MAO (in mitochondria) R R’ OH H Norepi OH CH 3")
Monoamine Oxidase (MAO) MAO (in mitochondria) R R’ OH H Norepi OH CH 3 Epi H H Dopamine MAO inhibitors (e. g. , tranylcypromine) are useful in the treatment of depression Brain levels of dopamine and norepi. ; also serotonin Aldehyde dehydrogenase Urinary metabolite R=OH Vanillylmandelic acid (VMA) R=H Homovanillic acid (HVA) 12
 • Tyramine found naturally in several types of")
Tyramine MAO Tyramine ( blood pressure) • Tyramine found naturally in several types of cheese; also beer and red wine. • Tyramine intake can cause hypertensive crisis in persons taking a MAO inhibitor ( norepi release) 13
 COMT Active catecholamine SAM S-Adenosylhomocysteine Inactive metabolite • COMT found in")
Catechol-O-Methyl Transferase (COMT) COMT Active catecholamine SAM S-Adenosylhomocysteine Inactive metabolite • COMT found in cytoplasm • Terminates activity of catecholamines • Catecholamine excretion products result from combined actions of MAO and COMT • Inhibitors of COMT (e. g. , tolcapone) useful in Parkinson’s disease 14
 Highly colored")
Melanin Formation Tyr hydroxylase Tyrosine O 2 DOPA Tyrosinase Melanin (Black polymer) Highly colored polymeric intermediates Melanin formed in skin (melanocytes), eyes, and hair In skin, protects against sunlight Albinism: genetic deficiency of tyrosinase Dopaquinone 15

Tryptophan catabolism Tryptophan has complex catabolic pathway: 1. the indol ring is ketogenic 2. the side chain forms the glucogenic products Kynurenate and xanthurenate are excrete in the urine.
 5 -Hydroxytryptophan CO 2 5")
Indole ring Trp hydroxylase Decarboxylase O 2 Tryptophan (Trp) 5 -Hydroxytryptophan CO 2 5 -Hydroxytryptamine (5 -HT); Serotonin MAO Dehydrogenase B 3 5 -Hydroxyindole acetic acid (5 -HIAA) (Urine)

Serotonin Metabolism: Melatonin 2 Steps Serotonin Melatonin: • Formed principally in pineal gland • Synthesis controlled by light, among other factors • Induces skin lightening • Suppresses ovarian function • Possible use in sleep disorders

Formation of Serine Glucose Dehydrogenase Glycolysis NAD+ NADH + H+ 3 Steps Pyruvate 3 -Phosphoglycerate 3 -Phosphohydroxypyruvate Inhibits Glutamate Transaminase a-Ketoglutarate Phosphatase Serine (Ser) 3 -Phosphoserine 19
 Serine hydroxymethyl")
Conversion of Serine to Glycine Folate Dihydrofolate reductase Serine Tetrahydrofolate (FH 4) Serine hydroxymethyl transferase (PLP-dep. ) Key intermediate in biosynthesis of purines and formation of thymine Glycine N 5, N 10 -Methylene FH 4 Important in biosynthesis of heme, porphyrins, and purines 20
 + FH 4 L-Homocysteine")
Sulfur-Containing Amino Acids Methionine Synthase (Vit. B 12 -dep. ) + FH 4 L-Homocysteine Methionine (Essential) Cystathionine b-synthase (PLP-dep. ) + 5 -Methyl FH 4 Serine Cystathionine lyase + b-Hydroxybutyrate Cysteine (Non-essential) Cystathionine 21

Methionine Metabolism: Methyl Donation S-Adenosyl methionine synthase Methionine ATP SAM Decarboxylase Decarboxylated SAM S-Adenosyl Methionine (SAM) CO 2 R-H Methyltransferases + R-CH 3 S-Adenosyl homocysteine 22
 (PLP-dep. ) Ornithine (from urea cycle) Putrescine CO 2")
Polyamine Biosynthesis Ornithine decarboxylase (ODC) (PLP-dep. ) Ornithine (from urea cycle) Putrescine CO 2 Decarboxylated SAM Spermidine synthase Spermine 5’-Methylthioadenosine Decarboxylated SAM Spermidine 23

Polyamines • Spermidine and spermine found in virtually all procaryotic and eucaryotic cells • Precise role undefined • Bind to nucleic acids • Inhibition of biosynthetic pathway: a-Difluoromethylornithine (DFMO) (Eflornithine) - inhibits ODC; used to treat Pneumocystis carinii infectons 24
 Arginine Creatinine (Urine) Glycine Ornithine Guanidoacetate Methyltransferase (Liver)")
Creatine and Creatinine Arginine-glycine transamidinase (Kidney) Arginine Creatinine (Urine) Glycine Ornithine Guanidoacetate Methyltransferase (Liver) SAM + ATP S-Adenosylhomocysteine + ADP Non-enzymatic (Muscle) Creatine kinase (Muscle) Creatine ATP ADP + Pi Phosphocreatine 25

Creatine and Creatinine Creatine: • Dietary supplement • Used to improve athletic performance Creatinine: • Urinary excretion generally constant; proportional to muscle mass Creatinine Clearance Test: • Compares the level of creatinine in urine (24 hrs. ) with the creatinine level in the blood • Used to assess kidney function • Important determinant in dosing of several drugs in patients with impaired renal function 26

Glycine biosynthesis from serine Reaction involves the transfer of the hydroxymethyl group from serine to the cofactor tetrahydrofolate (THF), producing glycine and N 5, N 10 -methylene-THF. Copy from: http: //themedicalbiochemistrypage. org/amino-acid-metabolism. html

Glycine oxidation to CO 2 Glycine produced from serine or from the diet can also be oxidized by glycine decarboxylase (also referred to as the glycine cleavage complex, GCC) to yield a second equivalent of N 5, N 10 -methylene-tetrahydrofolate as well as ammonia and CO 2. Copy from: http: //themedicalbiochemistrypage. org/amino-acid-metabolism. html

Cysteine and methionine are metabolically related The sulfur for cysteine synthesis comes from the essential amino acid methionine. SAM Condensation of ATP and methionine yield S-adenosylmethionine (SAM) SAM serves as a precurosor for numerous methyl transfer reactions (e. g. the conversion of norepinephrine to epinenephrine).

Cysteine synthesis Conversion of homocysteine back to Met. N 5 methyl-THF is donor of methyl group. * *folate + vit B 12 1. Conversion of SAM to homocysteine. 2. Condensation of homocysteine with serine to cystathione. 3. Cystathione is cleavaged to cysteine. Copy from: http: //themedicalbiochemistrypage. org/amino-acid-metabolism. html

Homocystinuria Genetic defects for both the synthase and the lyase. Missing or impaired cystathionine synthase leads to homocystinuria. High concentration of homocysteine and methionine in the urine. Homocysteine is highly reactive molecule. Disease is often associated with mental retardation, multisystemic disorder of connective tissue, muscle, CNS, and cardiovascular system.

Cysteine catabolism
 a-keto-b-methylbutyrate a-ketoisokaproate")
Catabolism of branched amino acids valine isoleucine a-ketoglutarate a-ketoisovalerate leucine glutamate (transamination) a-keto-b-methylbutyrate a-ketoisokaproate NAD+ oxidative decarboxylation Dehydrogenase of a-keto acids* CO 2 isobutyryl Co. A a-methylbutyryl Co. A NADH + H+ isovaleryl Co. A Dehydrogenation etc. , similar to fatty acid b-oxidation propionyl Co. A acetyl Co. A + acetoacetate
 (because of the characteristic")
Branched-chain aminoaciduria Disease also called Maple Syrup Urine Disease (MSUD) (because of the characteristic odor of the urine in affected individuals). Deficiency in an enzyme, branched-chain α-keto acid dehydrogenase leads to an accumulation of three branchedchain amino acids and their corresponding branched-chain α-keto acids which are excreted in the urine. There is only one dehydrogenase enzyme for all three amino acids. Mental retardation in these cases is extensive.
 ARG: can be made, but not enough")
Essential & Non-essential AA �Conditionally essential (i) ARG: can be made, but not enough (ii) HIS: controversial (essential for growth in children) (iii) PHE essential, TYR can be made from PHE but when enzyme is missing (phenyl- ketonuria) then PHE > TYR; Therefore TYR is essential (iv) MET�CYS; Similarly, if MET > CYS then CYS essential Even with excess, important in excretion NH 4+ therefore continue to be made

Serine biosynthesis from glycolytic intermediate 3 -phosphoglycerate Copy from: http: //themedicalbiochemistrypage. org/amino-acid-metabolism. html

Formation of alanine

Asparagine synthetase reaction

The glutamate dehydrogenase

Ammonia transport in the form of glutamine Excess ammonia is added to glutamate to form glutamine. Glutamine synthetase Glutamine enters the liver and NH 4+ is liberated in mitochondria by the enzyme glutaminase. Ammonia is remove by urea synthesis.

Biosynthesis of proline from glutamate

Conversion of methionine to propionyl-Co. A.

The phenylalanine hydroxylase reaction

Conversion of Amino Acids to Specialized Products
 DOPA decarboxylase Epinephrine (Adrenaline)")
Catecholamine Biosynthesis Catechol Tyr hydroxylase O 2 Tyrosine Dihydroxyphenylalanine (DOPA) DOPA decarboxylase Epinephrine (Adrenaline) Methyl transferase S-Adenosylhomocysteine SAM DOPA, dopamine, norepinephrine, and epinephrine are all neurotransmitters CO 2 Dopamine hydroxylase Dopamine Norepinephrine
 Arginine Creatinine (Urine) Glycine Ornithine Guanidoacetate Methyltransferase (Liver)")
Creatine and Creatinine Arginine-glycine transamidinase (Kidney) Arginine Creatinine (Urine) Glycine Ornithine Guanidoacetate Methyltransferase (Liver) SAM + ATP S-Adenosylhomocysteine + ADP Non-enzymatic (Muscle) Creatine kinase (Muscle) Creatine ATP ADP + Pi Phosphocreatine

β-Alanyl Dipeptides

Biosynthesis of hippurate

Polyamines: Conversion of spermidine to spermine

Nitric Oxide

glutathione

Carnitine GABA

Protein Metabolism
- Slides: 53