Metabolism of Amino Acid Section Physiological function and

Metabolism of Amino Acid

SectionⅠ Physiological function and nutritional value of protein 1. Physiological functions of protein Ø can not be replaced by carbohydrates or lipids 2. Nitrogen balance 3. Nutritional value of proteins: quantity + quality
: * Can not be synthesized by human body")
♣ Essential amino acids (8 kinds): * Can not be synthesized by human body * Must be supplied in the diet ♣ Non-essential amino acids(12 kinds) Nutritional value of proteins is related to the ratio of essential amino acids and non-essential amino acids
 Made by simple reactions")
Made by complex routes (in bacteria & plants) Made by simple reactions

Section II Digestion, absorption and putrefaction of proteins

I. Digestion of Dietary Protein Self-activation of pepsinogen to active pepsin pancreas Endopeptidase Exopeptidase Trypsin Chymotrypsin Elastase Carboxypeptidase A、B Dipeptidase

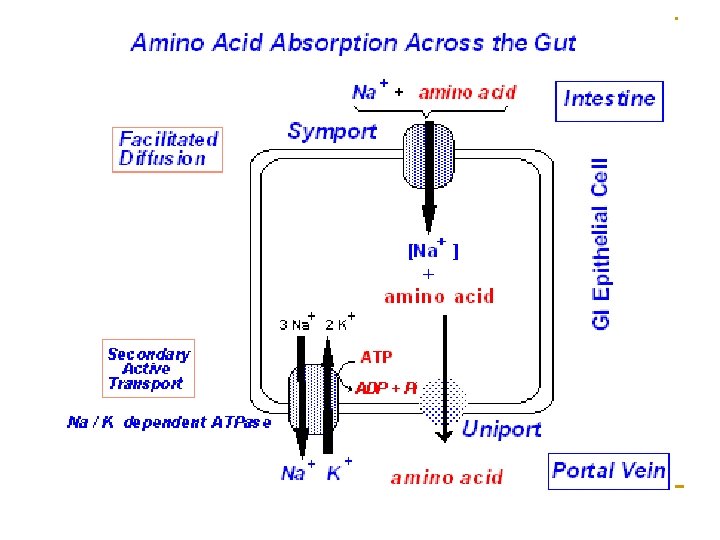
II. Absorption of amino acid Ø Site: small intestine Ø Mechanism 1. Na+-dependent carrier proteins: Different carriers involve in absorption of different amino acids. 2. γ-glutamyl cycle

γ-glutamyl transferase γ-glutamyl cycle

III. Putrefaction of protein The decomposition of the unabsorbed proteins, peptides or amino acids by anaerobic bacteria in the large intestine is termed putrefaction. n The resulting products are mostly toxic (amine, ammonia, phenol, indole and sulfureted hydrogen), most of which are excreted in feces.
 Formation of amines • Pr AAs amines (His, Lys, Trp, Tyr, Phe) co")
(1) Formation of amines • Pr AAs amines (His, Lys, Trp, Tyr, Phe) co 2 • producing false neurotransmitters. (2) Formation of ammonia • AAs deaminase NH + keto acid 3 • blood urease NH 3 (3) Others • tryptophan → indole and methylindole. • cysteine → H 2 S, Phenol etc.
 Phenylethylatamine 苯乙醇胺 Tyr CO 2 tyramine (0 H)")
Phe CO 2 phenylethylamine (0 H) Phenylethylatamine 苯乙醇胺 Tyr CO 2 tyramine (0 H) β- hydrotyramine β-羟酪胺(鱆胺)

Section Ⅲ General Metabolism of Amino Acids Sources of amino acids * Dietary proteins (the main source for our body) - digestion & absorption * Tissue proteins - degraded by proteolytic enzymes in lysosome - degraded by ubiquination in cytoplasm * Synthesis of non-essential AA in vivo

Ⅰ. Degradation of Tissue Proteins Protein Turnover: The process of continuous degradation and resynthesis of proteins. Proteins differ significantly in their turnover rates, which are measured in half-lives. Significances: Ø Metabolic flexibility(灵活性): key regulatory enzymes, peptide hormones, receptor molecules; Ø Protects cells from the accumulation of abnormal proteins; Ø Numerous physiological processes are dependent on timely degradative and synthetic reactions: eukaryotic cell cycle control and antigen presentation.

1 -8

n Pathway of tissue proteins degradation Ø Protein degradation pathway in lysosomes n occurs in lysosomes n ATP independent n Non-specific protein degradation for extracellular proteins membrane proteins long half-life intracellular proteins

n Pathway of tissue proteins degradation Ø Ubiquitin—proteasome degradation pathway n occurs in cytosol/nucleus n ATP dependent n Specific protein degradation for abnomal proteins short life proteins

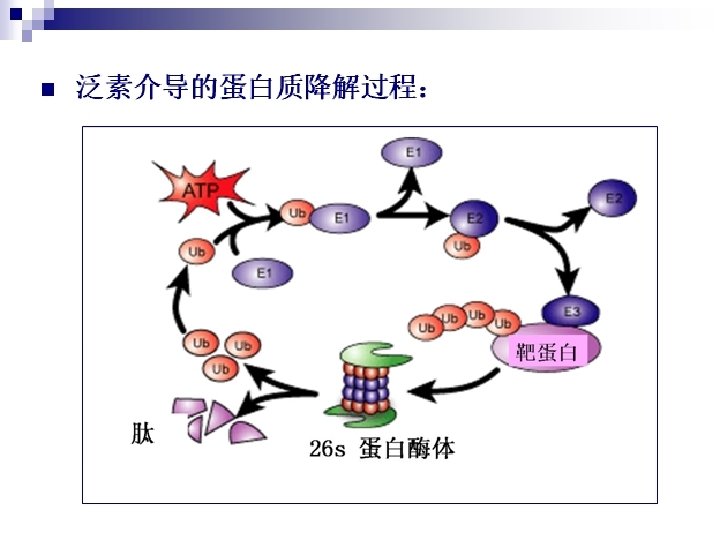
Ubiquitination * Ubiquitin: 8. 5 KD small protein of 76 aas * 泛素活化酶(ubiquitin activating enzyme, E 1) * 泛素结合酶(ubiquitin coupling enzyme, E 2) * 泛素-蛋白连接酶(ubiquitin-protein ligase, E 3) * 蛋白酶体(proteasome)- huge complex

ubiquitination Active Pr

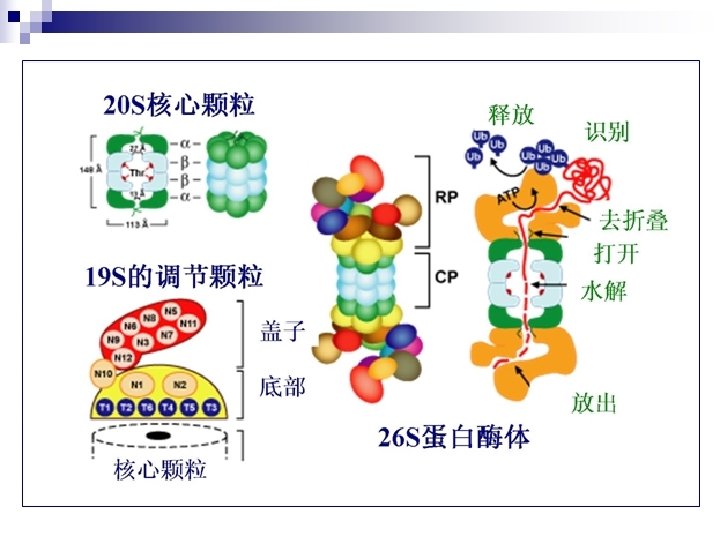
n Proteasome:huge complex n Proteasome exist in cell nuclear and cytoplasm, degrade abnormal and short-life proteins. 20 S core particle (CP) 26 s protein complex 2α loop: 7 subunits/loop 2β loop: 7 subunits/loop 19 S regulatory particles (RP): 18 subunits, 6 subunits have ATPase activity.

Ⅱ. Amino acid metabolic pool Tissue protein Dietary protein Tissue protein Amino acid metabolic pool Other pathways: Nitrogen-containing Compounds, one-carbon units, etc. Synthesis of Nonessential amino acids Sources and fates of amino acids

Fates of amino acids excretion from urine ammonia urea in liver *deamination non-essential aa α-ketoacid CO 2 +H 2 O + ATP Glc or lipids amine non-essential aa *decarboxylation CO 2

Ⅲ. Deamination of amino acids Amino acids R –C– COOH NH 2 R –C– COOH O ammonia

The first step in the catabolism of AAs is removing of their α-amino group. Four types of deamination: 1. Transamination 2. L-Glu oxidative deamination 3. Associated deamination* -transamination associated with oxidative deamination -transamination associated with purine nucleotide cycle 4. Amino acid oxidase deamination
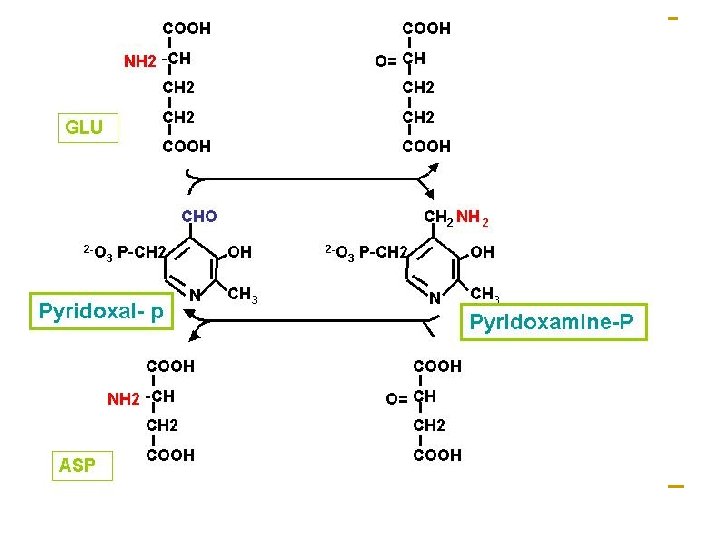
 Coenzyme: Pyridoxyl phosphate (PLP) (Vit. B 6)")
1. Transamination (Transaminase) Coenzyme: Pyridoxyl phosphate (PLP) (Vit. B 6)

• Distribute widely • Specificity of aminotransferases • Reversible reaction: • degradation of amino acids • amino acids synthesis • Can not remove free ammonia from molecular

The most important aminotransferases are to catalyze amino transfer between L-Glu and αketoacid:
 / glutamate-pyruvate transaminase (GPT)")
n Alanine aminotransferases (ALT) / glutamate-pyruvate transaminase (GPT)
 / glutamate-oxaloacetate transaminase (GOT) COOH Asp HC oxaloacetate NH 2")
n Aspartate aminotransferase (AST) / glutamate-oxaloacetate transaminase (GOT) COOH Asp HC oxaloacetate NH 2 α- ketoglutarate CH 2 COOH C=O CH 2 COOH AST glutamate

The activities of ALT and AST in different normal adult tissues Tissue AST ALT (unit/g wet tissue) Heart 156000 7100 Liver 142000 44000 Muscle 99000 4800 Kidney 91000 19000 Pancreas 28000 2000 Spleen 14000 12000 Lung 10000 700 Serum 20 16

Diagnosis value of serum aminotransferases Two important enzymes in the clinical diagnosis of human disease are serum AST and ALT- abundant in heart and liver (intracellular enzymes). They are released as part of the cell injury that occurs in myocardial infarction, infectious hepatitis, or other damage to either organ. Assays of these enzyme activities in blood serum can be used both in diagnosis and in monitoring the progress of a patient during treatment.

Pyridoxal phosphate-derived from vitamin B 6, is their coenzyme of transaminase serving as transporter amino group in the reaction.

The functional part of pyridoxal phosphate is an aldehyde functional group attached to a pyridine ring. The mechanism of transamination

2. L-Glu oxidative deamination ♣ L-Glutamate dehydrogenase Reversible n active in most tissue except skeletal and cardiac muscles; n allosteric enzyme (ATP/GTP inhibits, ADP/GDP activates).
 amino acid a-ketoglutarate")
3. Transamination associated with oxidative deamination (Associated deamination in most tissues) amino acid a-ketoglutarate Transaminase a-keto acid NH 3+NADH+H+ glutamate dehydrogenase glutamate H 2 O+NAD+ Degradation of amino acids

amino acid a-ketoglutarate Transaminase a-keto acid NH 3+NADH+H+ glutamate dehydrogenase glutamate H 2 O+NAD + Synthesis of nonessential amino acids

4. Transamination associated with purine nucleotide cycle in skeleton muscles and other tissues
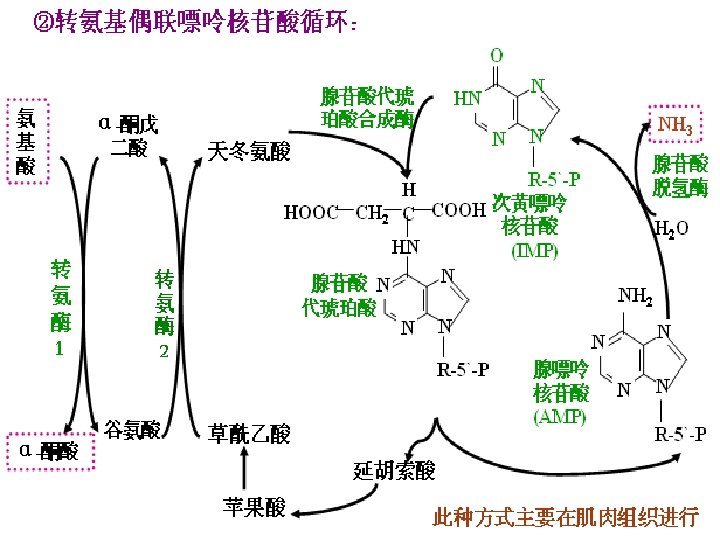
")
5. Oxidative deamination of AAs ♣ (In kidney and liver cells)

deamination NH 3 O R-CH-COOH α-ketoacid

Ⅱ. Fates of the carbon atoms of amino acids after deamination: n Be oxidized to produce CO 2+H 2 O+ATP via citric acid cycle and oxidative phosphorylation. n Be converted into non-essential amino acids by amination. n Be converted into glucose, lipids or ketone bodies. glucogenic amino acids ketogenic and glucogenic amino acids

glucose Ketone bodies

SectionⅣ Metabolism of Ammonia n Ammonia is extreme toxic compound to central nervous system. n The major way for detoxification of ammonia is to be converted into water-soluble, nontoxic urea in the liver, which is mainly passed via the bloodstream to the kidneys and excreted in the urine.

Ⅰ. Sources of ammonia in the body: 1. Deamination of amino acids and amine. 2. Large quantity of ammonia is produced in intestine. (1) putrefaction of dietary protein (2) hydrolysis of urea: blood urea diffuse into intestine, degraded by bacterial urease. The ammonia is re-absorbed into the blood → liver. NH 4+ (ammonium ion) < == > NH 3 + H+ (ammonia) So alkaline soapsuds can NOT be used in clinical clyster for victim with hyperammonemia. 3. Reabsorbed from kidney in which ammonia is produced by hydrolysis of glutamine by glutaminase glutamate + NH glutamine 3

Blood Gln cell Gln glutaminase Glu NH 3 urine NH 3 Glu NH 3 Acidification of urine NH 3 H+ NH 4+

Ⅱ. Transportation of ammonia The ammonia need to be transported from the generated tissue to the liver in a nontoxic form. 1. Alanine-Glucose cycle: • To transport ammonia in the nontoxic form of alanine from muscle to liver. • To regulate blood glucose indirectly and supply available glucose for muscle.

2. Transportation of ammonia with glutamine: Glutamine is a major transport form of ammonia. COOH In brain or muscle ADP+Pi Gln ATP synthetase CONH 2 CH NH 2 + NH 3 (CH 2) 2 COOH Glu CH NH 2 glutaminase In liver or kidney (CH 2) 2 COOH Gln In liver, NH 3 urea In kidney, NH 3 + H+ NH 4+ is excreted in the urine.

Transportation of ammonia
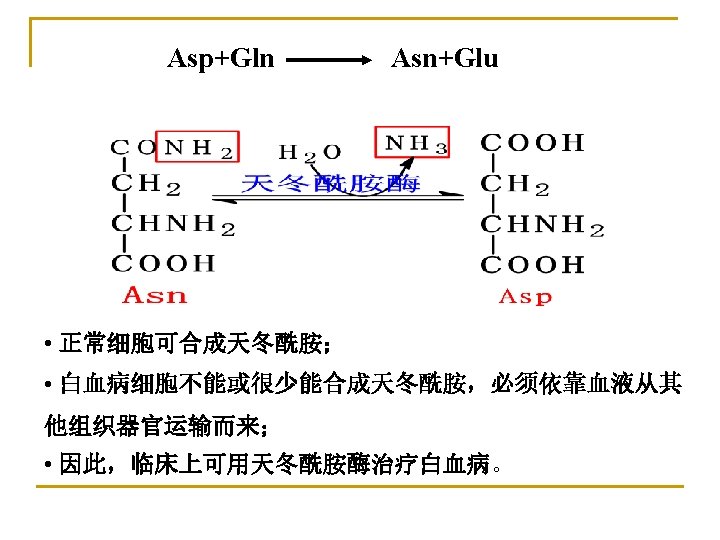

Ⅲ. Metabolic fates of ammonia 1. Synthesis of urea in liver 2. Synthesis of nonessential amino acids and nucleotides 3. Excretion from kidney as NH 4+

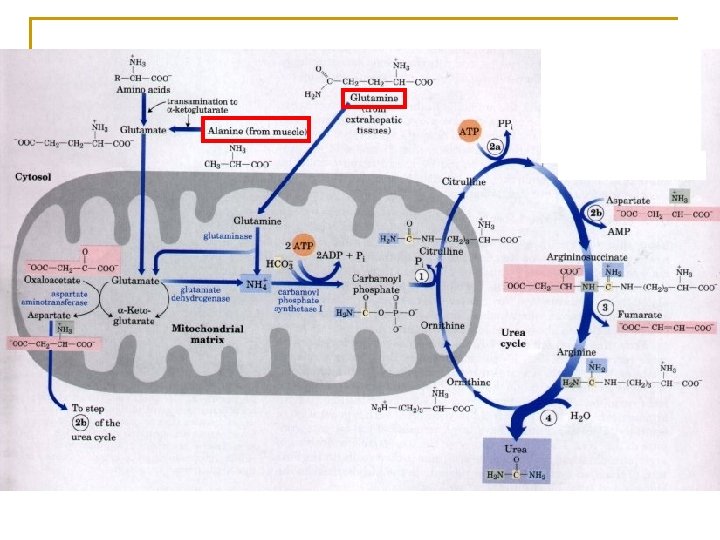
Ⅳ. Synthesis of Urea n Liver is the major organ for urea synthesis n Ornithine cycle (or urea cycle) Krebs and Henseleit 1932 n Formed from 2 NH 3 and CO 2 n The significance is to convert ammonia into urea that can be excreted from kidney and to detoxify ammonia

Process of urea cycle: n Formation of Carbamoyl phosphate n Formation of citrulline n Formation of arginine n Synthesis of urea • The first two steps occur in Mit. • Other steps occur in cytoplasm urea cycle

Formation of Carbamoyl phosphate n Occurs in the matrix of mitochondria. n Enzyme: carbamoyl phosphate synthetaseⅠ(CPS- I) n CPS- I is a regulatory enzyme (N-acetylglutamate, AGA as a positive modulator). n CPS-Ⅰis distinct from the cytosolic CPS-Ⅱ form, which has a separate function in pyrimidine biosynthesis.

Carbamoyl phosphate synthetase I ornithine transcarbamoylase TCA cycle
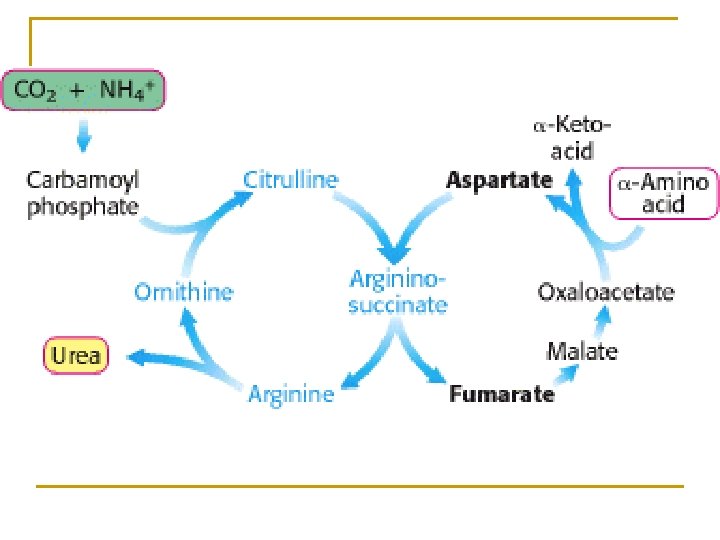
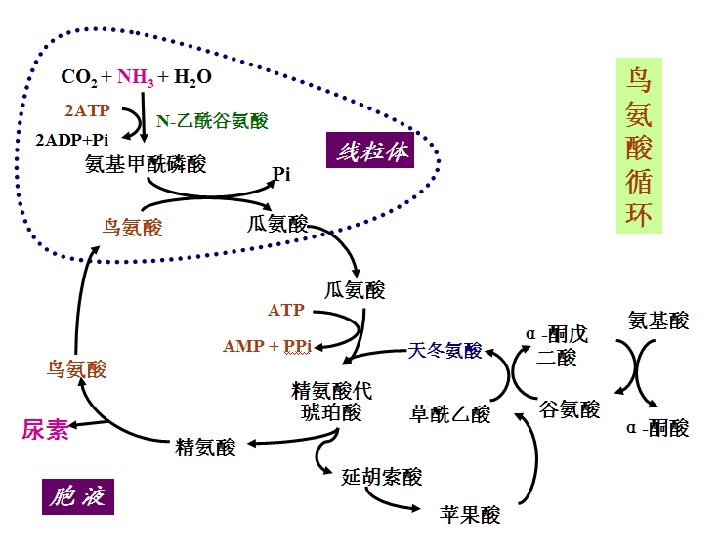
, CO 2 2. Process:")
Summary 1. Material: 2 NH 3 (free NH 3, Asp), CO 2 2. Process: first in MIT then in cytosol 3. Energy: The synthesis of one molecule of urea requires four high-energy phosphate groups. 2 NH 3 + CO 2 + 3 ATP + H 2 O urea+2 ADP+ AMP+4 Pi
")
Regulation of CPS- I Arginine (AGA)

Hyperammonemia and ammonia toxicosis The urea synthesis converts toxic ammonia to nontoxic urea. When liver function is damaged, urea synthesis will be blocked, then the blood ammonia levels increase. Hyperammonemia and hepatic coma and death will occur.

Hyperammonemia and ammonia toxicosis In brain Mechanism: n n n n Liver function ↓ Urea synthesis ↓ Blood ammonia ↑ Ammonia in brain↑ TCA in brain ↓ Energy for brain ↓ Brain function ↓ NH 3 ATP Glutamate glutamine NH 3

SectionⅤ Metabolism of Individual Amino Acids Ⅰ. Decarboxylation of amino acids R-CH-NH 2 decarboxylase RCH 2 NH 2 + CO 2 COOH pyridoxal phosphate (PLP) amine Amino acid Have important physiological functions Amine oxidase RCHO →RCOOH
 : inhibitory neurotransmitter Cys → taurine: a component of conjugated")
Glu →γ-aminobutyric acid (GABA) : inhibitory neurotransmitter Cys → taurine: a component of conjugated bile acid His → histamine: a mediator of inflammatory and allergic reaction Trp → 5 -hydroxytryptamine (5 -HT) inhibitory neurotransmitter and vasoconstrictor Polyamine: ornithine→putrescine→spermidine→spermine

Synthesis of histamine It is secreted by mast cells as a result of allergic and inflammatory reactions. Histamine is a powerful vasodilator
 Serotonin also called 5 hydroxytryptamine. • act as inhibitory neurotransmitter and")
serotonin (5 -HT) Serotonin also called 5 hydroxytryptamine. • act as inhibitory neurotransmitter and vasoconstrictor • Regulation of sleep, temperature and blood pressure.

polyamines Promoting the proliferation of cells SAM

Polyamines are intracellular polycationic molecules found in all living cells. They are essential for cell growth and differentiation. An elevated cellular polyamine concentration has been linked to the initiation and progression of cancer. Polyamine levels are higher in malignant cells and tumors compared to normal cells and tissues

Ⅱ. Metabolism of one-carbon units * One-carbon units are groups containing one-carbon atom produced by some amino acid metabolism. * Types: methyl(-CH 3) methenyl(-CH=) methylene (-CH 2 -) formyl (-CHO) formimino(-CH=NH) * Carrier(coenzyme): tetrahydrofolate (FH 4) * Serve as donors of one-carbon units in the biosynthesis of purine and pyrimidine.

Role of folic acid in amino acid metabolism One carbon unit can not exist freely, FH 4 serves as carrier to transfer the onecarbon units.
. Dihydrofolate (FH 4)")
Folic acid is a kind of water-soluble vitamin. (DHFR). Dihydrofolate (FH 4)

Formation of one carbon unit Ser、 Gly,His,Trp

Ser hydroxymethy Transferase Glycine lyase

formiminoglutamic aci

Interconvertion of one carbon units N 10 CHO FH 4 NH 3 N 5、N 10 = CH FH 4 NADPH+H N 5、N 10 N 5 CH 3 NADP N 5 CH = NH FH 4 CH 2 FH 4 The different forms of NADH+H FH 4 are interconvertible. NAD The formation of N 5 - FH 4 methyl - FH 4 is inreversible reaction.

Significances of one-carbon unit metabolism n n n As the precursors for purine and pyrimidine synthesis Links amino acid metabolism with nucleotide metabolism. Deficiency of folic acid or block of one-carbon unit metabolism can influence DNA and RNA synthesis. The victim may get megaloblastic anemia. 巨幼红细胞性贫血

Ⅲ. Metabolism of sulfur-containing amino acids 1. Metabolism of methionine ① Transmethylation * active methyl group, which can be transferred to a variety of acceptor molecules (50 sorts) (SAM)
 is a powerful methylating involve in: - Ethanolamine→choline - Capping")
• S-Adenosylmethionine (SAM) is a powerful methylating involve in: - Ethanolamine→choline - Capping of m. RNA SAM R - Methylation of DNA - Methyl modification of enzymes A variety of methyl transferases - Synthesis of creatine, epinephrine, carnitine - Methylation in biotransformation R-CH 3 S-Adenosylhomocysteine
 ② Methionine cycle")
Met SAM FH 4 Methyl transferase/ methionine synthase (Vit B 12) ② Methionine cycle methyltransferase N 5 -CH 3 FH 4 hydrolase homocysteine S-adenosyl homocysteine

Homo-Cys N 5 -CH 3 FH 4 N 5, N 10 CH 2 -FH 4 One-carbon metabolism Met cycle FH 4 Gly Ser Met ATP SAH R-CH 3 R SAM PPi +Pi

Significance of Met cycle n The importance of Met cycle is to renew Met from onecarbon units for its reuse. n The reaction for generation of N 5 -CH 3 -FH 4 is virtually irreversible. Methionine synthase is the only mammalian enzyme known to act on N 5 -CH 3 -FH 4. Deficiency of Vit B 12 may reduce the enzyme activity, lead to accumulation of N 5 -CH 3 -FH 4, and decrease functional FH 4 level, which is insufficient for synthesis of nucleotides. The victim with the deficiency of Vit B 12 may get megaloblastic anemia.
")
(Vit. B 6)

③ Creatine synthesis Creatine phospate is a high-energy compound that can reversibly donate a phosphate group to ADP to form ATP. CK Isozymes: MM type BB type MB type

In liver

Met is glucogenic amino acid glucose

2. Metabolism of cysteine and cystine ① Interconversion of cysteine and cystine 2 –SH -2 H +2 H -S-S- ② Metabolism of sulfate ATP + SO 42 - → 3’-PO 3 H 2 - AMP – SO 3 - (PAPS) is called active sulfate, which serves as sulfate donor in the sulfurylation reaction (biotransformation in liver, aminoglucose).

Ⅳ. Metabolism of aromatic amino acids 1. Metabolism of phenylalanine and tyrosine (Phe and Tyr) Phe * Tyr hydroxylase Phe hydroxylase Tyr Dopa transamination CO 2 tyrosinase Dopa Phenylpyruvate 肾上腺髓质 melanin Acetoacetate + fumarate ketone bodies glucose Dopamine (in brain) Norepinephrine SAM Epinephrine

Phe Tyrosine gives rise to a family of catecholamines that includes: dopamine, norepinephrine, epinephrine. catecholamines

albinism n Albinism disease is due to a inborn deficiency in tyrosinase, so melanin can not be synthesized. n Melanin is a pigment that occurs in many tissues particularly in the eye, hair, and skin. . . The patients have light color in tissues and they are photosensitive.
 v Deficiency in Phe hydroxylase leads to PKU. Since patients cannot")
Phenyl ketonuria (PKU) v Deficiency in Phe hydroxylase leads to PKU. Since patients cannot convert Phe to Tyr, the phenylpyruvates are accumulated in blood and excreted in the urine. v The PKU is characterized as mental deficiency. The brain of newborn infants can be injured by PKU. v PKU can be prevented by a diet control with low levels of Phe.

2. Metabolism of Trp Pyruvate→Glucose Acetoacetyl Co. A→ Ketone bodies N 10 -CHO-FH 4 (5 -HT)

Ⅴ. Metabolism of branched amino acids l Val →succinyl Co. A → glucose (glucogenic amino acids) l Leu → acetyl Co. A + acetoacetyl Co. A→ ketone bodies (ketogenic amino acids) l Ile → acetyl Co. A+ succinyl Co. A → glucose & ketone bodies (ketogenic and glucogenic amino acids)

Key Terms Ø Essential amino acids Ø Putrefaction of protein Ø One-carbon units
- Slides: 95