Metabolic Acidosis and Congenital Diarrhea Dr Amir Bar

Metabolic Acidosis and Congenital Diarrhea Dr. Amir Bar, Bnei-Zion Medical Center, Haifa


 : מעבדה - ג. ע Na+ BE HCO PCO 3 2 p. H")
(3) : מעבדה - ג. ע Na+ BE HCO PCO 3 2 p. H 151 06: 43 18. 12 144 -21. 5 8. 2 25. 7 7. 11 20: 11 18. 12 147 -19. 4 8. 6 21. 0 7. 21 22: 07 18. 12 139 -20. 0 8. 2 20. 7 7. 20 01: 45 19. 12 137 -17. 5 9. 5 20. 3 7. 27 09: 57 19. 12


Metabolic Acidosis 1. Normal AG Bicarbonate loss v Renal – RTA v GI – diarrhea 2. (AG = 16) Increased AG Additional anion v Sepsis – lactate v Diabetes – ketones v Inborn errors of metab. (Normal WBC & CRP, Normal lactate) (Normal glucose levels, No ketonuria) (No hyperammonemia, Negative organic & amino Ac)
 v Normal AG, hyperchloremic metabolic acidosis resulting from either impaired")
Renal tubular Acidosis (RTA) v Normal AG, hyperchloremic metabolic acidosis resulting from either impaired HCO 3 reabsorption or impaired H+ excretion v Type 1 v Type 2 v Type 3 v Primarily in Pt with inherited carbonic anhydrase def v Type v (Distal) (Proximal) (Mixed of type 1 and 2) 4 Hyperkalemic, deficiency/resistance to
 § § § Sporadic Hereditary (AD, AR) Fanconi")
Proximal RTA: dd § Isolated (Rare) § § § Sporadic Hereditary (AD, AR) Fanconi syn (more common) glycosuria, aminoaciduria, proteinuria (LMW), phosphaturia § Primary § § Sporadic Hereditary (AD, AR) § Cystinosis, Lowe syndrome, Galactosemia, Tyrosenemia, Fructosemia, Fanconi-Bickel syndrome, Wilson disease, Mitochondrial diseases § Secodary: § Heavy metals, Outdated tetracycline, Gentamicin, Ifosfamide, Cyclosporine, tacrolimus

Proximal Tubule HCO 3 - Peritubular fluid Na+ Na+ HCO 3 - H+ H 2 CO 3 H+ K+ H 2 O Carb. Anhyd. H 2 O+CO 2+HO- Na+ 85% HCO 3 - Carb. Anhyd. HCO 3 -
 Serum HCO 3 levels were in the range of 7 -12 Proximal RTA:")
(A) Serum HCO 3 levels were in the range of 7 -12 Proximal RTA: Dx v Hyperchloremic/Hypokalemic metabolic acidosis v The serum HCO 3 level falls until it reaches the PT-HCO 3 threshold (15 -16) >> urine excretion stops v Urine is acidified (p. H < 5. 5) when serum HCO 3 < 16 (Normal distal H+ secretion) >> RTA-2, while alkaline urine implies RTA-1 v HCO(C) serum 3 provision Proximal –RTA is HCO 3 will (B)be HCO 3=23 rarely isolated increased but not !!! to the normal range, and
 Secondary v")
Distal RTA: dd v Primary v v v Sporadic Hereditary (AD, AR) Secondary v Interstitial nephritis l l l l l v Obstructive uropathy Vesicoureteral reflux Pyelonephritis Transplant rejection Sickle cell nephropathy Ehlers-Danlos syndrome Lupus nephritis Nephrocalcinosis Medullary sponge kidney Hepatic cirrhosis Toxins/Medications l l Amphotericin B Lithium Toluene Cisplatin

Distal Tubule Collecting duct Peritubular fluid Na+ H+ H+ H 2 O Cl 15% CO 2+HO- H+ + NH 3 Cl-/NH 4+ HCO 3 - HCO 3 NH 3
 Ur: Na-20, K-8. 8, Cl 50. 6 >> Ur-AG =")
Distal RTA: Dx (A) Ur: Na-20, K-8. 8, Cl 50. 6 >> Ur-AG = ( 21. 8) v Hyperchloremic/Hypokalemic M. A. v “Urine AG” (Na+ + K+ - Cl-): v Normal subjects: Met. Ac. >> acidification of the urine via NH 4/Cl excretion (only the Cl is presence in the equation) >> Negative “Urine AG” v Low urine p. H v v Abnormal urine acidification, low NH 4/Cl excretion >> zero or positive Urine AG and high urine p. H (B) Urine p. H=5. 0

Metabolic Acidosis v Increased AG § Additional anion v v v Sepsis – lactate Diabetes – ketones Inborn errors of metab. v Normal AG § Bicarbonate loss v v Renal – RTA GI – diarrhea (Normal WBC & CRP, Normal lactate) (Normal glucose levels) (No hyperammonemia, Negative organic & amino Ac) (AG = 16)

Diarrhea: Pathophysiology 1. 2. 3. 4. 5. Secretory diarrhea Osmotic diarrhea Ion transport defects Reduced surface area Abnormal motility

Hypochloremia/Hyponatremia/Alkalosis Secretory diarrhea HCO 3 - H+ Villus Epithelial cell Na+ Cl- - • c. AMP • c. GMP GI Lumen • Ca+2 + Cl. Crypt Epithelial cell K+ Na+ K- Na+-K+/Cl

Osmotic diarrhea Osmotic Secretor y Stool volume <200 ml/d >200 ml/d Fasting D. stops D. cont Stool Na+ <70 m. Eq/L >70 m. Eq/ L Stool p. H <5 >6 v. Red. Diarrhea Neocate (lactase def) Subs under Positive Negative v. Carbohydrate free diet (glucose-Galactose malabs. ) v. Diarrhea during NPO

Ions Transport Defects Hyponatremia Amino Ac Na+ H+ Glu Na+ Hyponatremia Na+ Bile Ac Cl- Na+ HCO 3 - Alkalosis

Diarrhea: Pathophysiology 1. 2. 3. 4. 5. Secretory diarrhea Osmotic diarrhea Ion transport defects Reduced surface area Abnormal motility
 v 1968 - 1 st described by the following")
Intractable Diarrhea of Infancy (IDI) v 1968 - 1 st described by the following features: 1. 2. 3. Diarrhea in an infant <3 m Lasting > 2 w 3 or more negative stool cultures

IDI / PDI: Causes v. The list of causes can be divided into: v. Normal villus-crypt axis v. Villus atrophy
 Ion transport defects v v Chloride-bicarbonate")
IDI / PDI: Causes A. Normal Villus a) Ion transport defects v v Chloride-bicarbonate exchanger (chloride-losing d. ) Sodium-hydrogen exchanger (congenital sodium d. ) Ileal bile acid receptor defect Sodium-glucose cotransporter (glucose-galactose mal) b) Micronutrient deficiency v Acrodermatitis enteropathica (zinc def) c) Enzyme deficiency v Enterokinase def d) Congenital short bowel
 v Tufting")
IDI / PDI: Causes B. Villus Atrophy v Microvillus inclusion disease (MVID) v Tufting enteropathy v Autoimmune enteropathy v IPEX syndrome v Infectious enteropathy v Post-infectious enteropathy v Allergic enteropathy v Idiopathic

Microvillus Inclusion Disease The 2 nd most common identified cause of IDI/PDI beginning in the 1 st week of life (After infection) v Various names: v a) Microvillus inclusion disease v (microvillus inclusions in enterocytes/colonocytes - the characteristic diagnostic feature on EM) b) Congenital microvillus atrophy c) Familial microvillous atrophy d) Davidson’s syndrome

Davidson’s syndrome 1978, Davidson et al - 5 newborns with severe, persistent diarrhea LM: thin mucosa, villous atrophy EM: intra-cytoplasmic cysts made up of brush border and increased secretory granules From this 1 st clinical and histologic description, MVID has been established as a distinct disease within the syndrome of IDI

MVID Typical form - 1 st days of life, a severe watery diarrhea (>250 -300 m. L/kg/d), which can be mistaken for urine Massive diarrhea >> Life-threatening >> dehydration, electrolyte imbalance, and metabolic acidosis within hours, persists despite GI rest Atypical clinical presentation - predominant occlusive syndrome “Late-onset” (>1 m), less severe diarrhea, secretory granules and microvillous inclusions are present, but distributed differently

MVID Crypt cells – increase in secretory granules, otherwise appear normal on EM, welldeveloped brush border In contrast, in mid- to upper villous – rare/absent microvilli, the diagnostic presence of microvillous inclusions The colon is involved, and although it may be easier to Bx the rectum, Dx features are not easily recognized

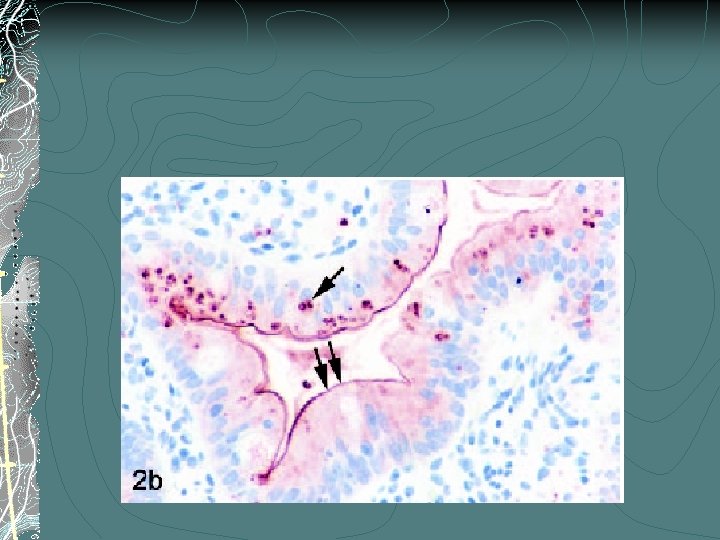
MVID: Histology Variable degree of villous atrophy, generally w/o any inflammatory infiltrate Staining: Periodic Acid Schiff (PAS) - positive secretory granules and abnormal brush border pattern CD 10

PAS-Control PAS-MVID CD 10 - control CD 10 - MVID
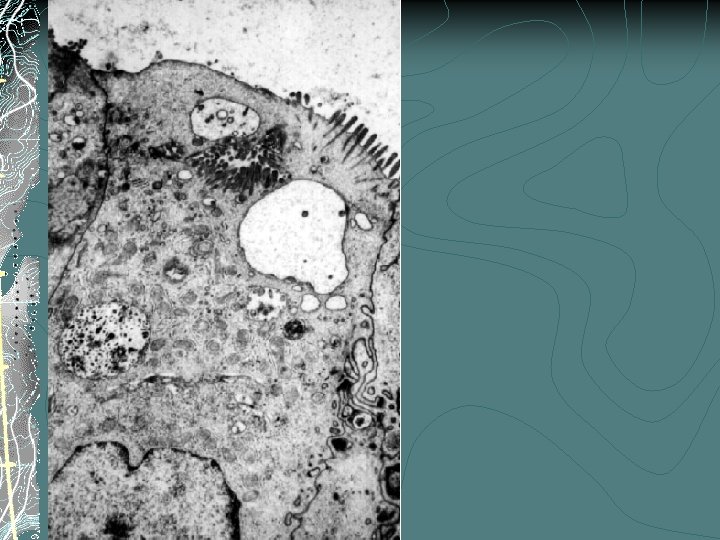

MVID: Pathogenesis A defect in the membrane trafficking of immature / differentiating enterocytes Enterocyte cytoskeleton Autosomal recessive Affected siblings Consanguinity No candidate genes have been identified

MVID: Tx It is recommended that once the diagnosis of typical MVID has been made, transplant should be considered Conversely, Pt with a late-onset or atypical MVID should not be automatically scheduled for transplant

Tufting Enteropathy/Intestinal epithelial Dysplasia v Chronic watery diarrhea on the 1 st few months v Dysmorphic features - in some affected infants v The long-term prognosis is variable

Tufting Enteropathy: Morphology – LM v The characteristic feature is the epithelial “tufts” (80 -90% of epithelial surface, in contrast to other known enteropathies <15%) + other typical findings: v Total or partial villus atrophy v Crypt hyperplasia v Normal or slightly increased density of inflammatory cells in the lamina propria v No colonic involvement

Tufting Enteropathy

Tufting Enteropathy: Pathogenesis v The molecular basis for TE is unknown v Defect in adhesion molecules ? v A genetic defect ? ? v Cluster of patients in Malta v Involved families can have many affected infants

Autoimmune Enteropathy v Villus atrophy, infiltration of activated T cells into the lamina propria v In contrast to MVID and Tufting E. : v Extra-intestinal manifestations of autoimmunity (arthritis, Membranous GN, IDDM, hepatitis, hypothyroidism, hemolytic anemia, thronbocytopenia v Rarely had a family history of unexplained infantile diarrhea v Onset frequently > 2 m life v Responsive to immune suppression Tx

Autoimmune Enteropathy: Morphology v The histopathology is similar to celiac disease, except that there is a relative paucity of intraepithelial lymphocytes v Most of the affected infants have no history of gluten ingestion before the onset of diarrhea
 v Bx: total villus atrophy, crypt hyperplasia, crypt abscesses are")
Autoimmune Enteropathy: Morphology (Cont’) v Bx: total villus atrophy, crypt hyperplasia, crypt abscesses are identified in severely affected cases v The lesions are not confined to the small bowel; can be seen in the stomach and colon v Immunohistochemistry: increase in CD 3 positive lymphocytes within the epithelium and lamina propria

Autoimmune Enteropathy: Pathogenesis v Circulating systemic antibody against enterocytes v The villus atrophy and crypt hyperplasia are both considered 2 nd features of an autoimmune-induced injury to the gut v Immune suppression Tx: v Antibody levels decline/disappear v The titer may correlate with the volume of stool output

IPEX Syndrome v IPEX is characterized by: v Immune dysregulation v Polyendocrinopathy v Enteropathy v X-linkage v The syndrome has many intestinal manifestations in common with autoimmune enteropathy, including villus atrophy with a marked infiltration into the lamina propria of activated T cells

IPEX Syndrome v The genetic basis is a mutation of the FOXP 3 gene, a transcription factor involved in the proliferation of CD 4+ T cells v Autoimmune enteropathy v IDDM, thyroid disease, eczematous ichthyosis hemolytic anemia
 v Tufting")
IDI / PDI: Causes B. Villus Atrophy v Microvillus inclusion disease (MVID) v Tufting enteropathy v Autoimmune enteropathy v IPEX syndrome v Infectious enteropathy v Post-infectious enteropathy v Allergic enteropathy v Idiopathic

Thank You!
- Slides: 45