Mekanika Molekuler Pertemuan ke2 Pendahuluan Mekanika molekuler Molecular

 adalah pendekatan modeling berdasarkan mekanika klasik • Terminologi")
 yang dipilih, ada berapa banyak?")

 • Jika memilih inti atom dan elektron sebagai partikel")
 dari suatu sistem")

 Relative Strength Strong Interaction Weak Interaction Quarks, leptons")


")
 • Atom-atom dalam molekul disatukan bersama oleh ikatan kimia • Saat")

")


























: This reference sodium ion has two negative chloride")
: The attractive Coulomb energy due to the")










 Perfect Ag. Cl. (b) A Frenkel defect in Ag. Cl (a) (b) •")







- Slides: 62
Mekanika Molekuler Pertemuan ke-2
Pendahuluan • Mekanika molekuler (Molecular Mechanics) adalah pendekatan modeling berdasarkan mekanika klasik • Terminologi yang sama dengan pendekatan ini adalah Force Field Method • Satuan penyusun (building blocks) dalam metode mekanika molekuler adalah atom, elektron tidak dianggap sebagai partikel individual • Konsekuensinya ikatan antar atom tidak dilihat sebagai hasil penyelesaian persamaan Schrödinger untuk elektron • Informasi tentang ikatan dinyatakan secara eksplisit yang berarti diillustrasikan secara fisik bukan sebagai hasil interaksi elektron valensi • Mekanika molekuler telah terbukti bermanfaat untuk menjelaskan sistem dengan molekul besar atau padatan kristalin
Menguraikan Sistem • Deskripsi sistem: apa unit dasar (partikel) yang dipilih, ada berapa banyak? • Kondisi awal: Dimana posisi partikel dan bagaimana kecepatannya • Interaksi: Apa bentuk persamaan matematika untuk gaya yang bekerja antar partikel tersebut • Persamaan dinamik: Apa bentuk persamaan matematika yang sistem yang berubah dengan waktu?
Hierarki Satuan Penyusun untuk Mengurai Sistem Kimia Elektron Atoms Molekul Nuklei Quarks Protons Neutrons Makromolekul
Pemilihan Satuan Penyusun (Building Blocks) • Jika memilih inti atom dan elektron sebagai partikel penyusun, kita bisa mengurai atom dan molekul namun tidak bisa mengurai struktur internal inti atom • Jika memilih atom sebagai partikel penyusun, kita bisa mengurai struktur molekul namun tidak bisa mengurai distribusi elektron • Jika memilih molekul (asam amino) sebagai partikel penyusun, kita bisa mengurai struktur overall makromolekul (protein) namun tidak bisa mengurai pergerakan atom-atom dalam molekul.
Pemilihan Kondisi Awal • Posisi ruang yang lengkap (complete phase space) dari suatu sistem adalah sesuatu yang sangat besar: Mencakup semua nilai yang mungkin dari posisi dan kecepatan satu partikel • Kita hanya bisa mengurai sebagian kecil saja dari kondisi ini • Misalnya suatu isomer (struktural atau konformasional) dan reaksi kimia ingin coba diuraikan • Senyawa C 6 H 6 memiliki banyak kemungkinan struktur dan konformasi, namun jika kita spesifik pada senyawa benzene, maka kondisi awal sistem menjadi lebih sederhana
Interaksi Partikel dan Persamaan Dinamik • Pada level atomik, interaksi dasar yang bekerja hanya interaksi elektromagnetik • Pada pendekatan Mekanika Molekuler, interaksi antar partikel disusun dalam bentuk parameter yaitu interaksi stretching, bending, torsional, van der Waals dll. • Persamaan dinamik menjelaskan bagaimana suatu sistem berubah dengan perubahan waktu, Misal: dengan menggunakan GLBB kita bisa menjelaskan posisi sistem setelah waktu tertentu
Interaksi Fundamental Name Particles Range (m) Relative Strength Strong Interaction Weak Interaction Quarks, leptons 10 -15 100 0. 001 Electromagnetic Charged particles Mass particles 1 10 -40 Gravitational
Keterangan • Strong interaction adalah gaya yang menahan inti atom agar tetap utuh walaupun ada tolak menolak antar proton didalam • Weak interaction gaya yang bertanggung jawab pada peluruhan inti atom dengan mengkoversi neutron menjadi proton ( -decay) • Keduanya adalah gaya yang bekerja short range dan hanya signifikan within the atomic nucleus • Interaksi elektromagnetik dan gravitasional berbanding terbalik dengan jarak partikel • Interaksi elektromagnetik terjadi antara partikel bermuatan • Interaksi gravitasional terjadi antara partikel yang memiliki masa
Pendekatan didalam Force Field a. k. a Mekanika Molekuler • Force field menggunakan pendekatan mekanika klasik seperti persamaan Newton untuk mendeskripsikan sistem • Aspek kuantum dan energi elektron ditiadakan/tidak diperhitungkan • Dengan pendekatan klasik, permasalahan direduksi menjadi menentukan energi sistem pada struktur geometri tertentu • Seringkali juga digunakan untuk menentukan geometri untuk molekul yang paling stabil atau konformasi terbaik yang melibatkan interkonversi antar konformasi • Untuk keperluan ini perhitungkan diarahkan pada penentuan energi minima pada potential energy surface
Potential Energy Surface (Flash info)
Definisi (Flash info) • Atom-atom dalam molekul disatukan bersama oleh ikatan kimia • Saat atom terdistorsi, ikatan akan meregang atau menekuk/mengkerut yang menyebabkan energi potensial sistem meningkat • Setelah susunan geometri atom-atom yang baru terbentuk, molekul berada dalam kondisi stasioner. Pada posisi ini energi sistem tidak dipengaruhi oleh energi kinetik tetapi oleh posisi atom-atom (potensial) • Energi dari molekul merupakan fungsi dari posisi inti, saat inti bergerak, elektron secara cepat akan menyesuaikan • Hubungan antara energi molekuler dan geometri molekuler dapat dipetakan menjadi sebuah potential energy surface
Terminologi dalam Mekanika Molekuler • Molekul dalam MM diilustrasikan sebagai ball and spring dimana atom digambarkan memiliki ukuran dan kelembutan tertentu sedangkan ikatan digambarkan memiliki panjang dan kekakuan tertentu • Dasar dari pendekatan FF/MM ini adalah bahwa molekul tersusun atas unit dengan struktur yang serupa hanya berada dalam molekul yang berbeda • Misalnya semua ikatan ini sama pada molekul apa pun v. C H memiliki panjang 1, 06 sd 1, 10 Å v. Vibrasi regang C H 2900 sd 3300 cm-1 v. C H dapat dikembangkan lagi menjadi C H yang terikat pada ikatan tunggal, ganda atau rangkap 3
Tipe Atom dalam MM • Penggambaran molekul yang tersusun atas unit struktural (gugus fungsi) serupa dengan bentuk molekul yang berbeda pada Kimia Organik • Kimiawan organik biasanya menggunakan ball n stick atau huruf nama atom dan garis ikatan untuk menggambarkan molekul • FF method mirip dengan pendekatan ini dengan penambahan atom dan ikatan tidak memiliki satu ukuran dan panjang yang fixed • Unit struktural yang serupa pada molekul yang berbeda ini diimplementasikan dalam FF dengan istilah tipe atom • Tipe atom tergantung pada nomor atom dan jenis ikatan kimia yang terlibat • Dalam MM 2 ada 71 tipe atom yang berbeda
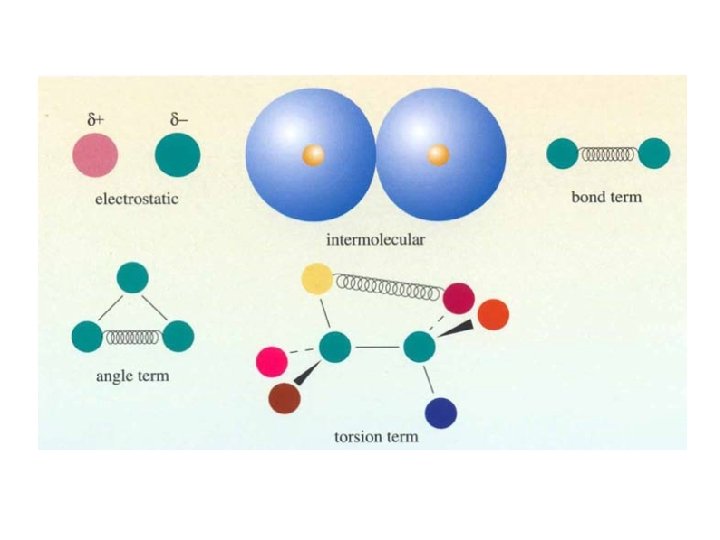
Energi dalam Force Field Method • Energi dalam Force Field ditulis sebagai jumlah dari semua suku • Masing-masing suku menguraikan energi yang dibutuhkan untuk mendistorsi molekul dalam arah tertentu EFF = Estr + Ebend + Etors + Evdw + Eel + Ecross • Dimana Estr adalah energi stretching ikatan antara 2 atom, Ebend energi yang dibutuhkan untuk membengkokkan sudut ikatan, Etors energi untuk proses rotasi memutar disekitar ikatan, Evdw dan Eel menguraikan interaksi atom-atom non-ikatan dan Ecross menguraikan kopling antar 3 suku awal diatas
Energi dalam Force Field
The Stretch Energy • Estr adalah fungsi energi untuk meregangkan ikatan antara 2 tipe atom A dan B • Dalam bentuknya yang paling sederhana, Estr dituliskan sebagai deret Taylor disekitar Panjang ikatan “natural” atau “kesetimbangan” R 0. • Parameter R 0 bukan Panjang ikatan kesetimbangan sembarang molekul, • Ia adalah parameter yang saat digunakan untuk menghitung struktur energi minimum suatu molekul akan menghasilkan geometri dengan Panjang ikatan kesetimbangan berdasarkan eksperiment
The Bending Energy • Ebend adalan energi yang dibutuhkan untuk membengkokan sudut yang dibentuk oleh 3 atom A B C, dimana ada ikatan yang terbentuk antara A dan B dan antara B dan C • Bentuk persamaannya juga merupakan deret Taylor disekitar sudut ikatan “natural” yang berakhir pada orde kedua dan memberikan pendekatan harmonik
The Out-of-Plane Bending Energy • If the central B atom in the angle ABC is sp 2 -hybridized, there is a significant energy penalty associated with making the centre pyramidal, since the four atoms prefer to be located in a plane. If the four atoms are exactly in a plane, the sum of the three angles with B as the central atom should be exactly 360°, however, a quite large pyramidalization may be achieved without seriously distorting any of these three angles. • Taking the bond distances to 1. 5Å, and moving the central atom 0. 2 Å out of the plane, only reduces the angle sum to 354. 8 (i. e. only a 1. 7° decrease per angle). • The corresponding out-of-plane angle, , is 7. 7 for this case. • Very large force constants must be used if the ABC, ABD and CBD angle distortions are to reflect the energy cost associated with the pyramidalization.
• This would have the consequence that the in-plane angle deformations for a planar structure would become unrealistically stiff. • Thus a special out-of-plane energy bend term (Eoop) is usually added, while the in-plane angles (ABC, ABD and CBD) are treated as in the general case above • Eoop may be written as a harmonic term in the angle (the equilibrium angle for a planar structure is zero) or as a quadratic function in the distance d, as given in equation below
The Out-of-Plane Bending Energy
The Torsional Energy • Etors describes part of the energy change associated with rotation around a B—C bond in a four-atom sequence A—B—C—D, where A—B, B—C and C —D are bonded • Looking down the B—C bond, the torsional angle is defined as the angle formed by the A—B and C—D bonds as shown in Figure. The angle may be taken to be in the range [0°, 360°] or [− 180°, 180°].
• The torsional energy is fundamentally different from Estr and Ebend in three aspects: 1. A rotational barrier has contributions from both the nonbonded (van der Waals and electrostatic) terms, as well as the torsional energy, and the torsional parameters are therefore intimately coupled to the non-bonded parameters. 2. The torsional energy function must be periodic in the angle : if the bond is rotated 360° the energy should return to the same value. 3. The cost in energy for distorting a molecule by rotation around a bond is often low, i. e. large deviations from the minimum energy structure may occur, and a Taylor expansion in is therefore not a good idea. • To encompass the periodicity, Etors is written as a Fourier series.
• The n = 1 term describes a rotation that is periodic by 360°, the n = 2 term is periodic by 180°, the n = 3 term is periodic by 120°, and so on. The Vn constants determine the size of the barrier for rotation around the B—C bond. • Depending on the situation, some of these Vn constants may be zero. In ethane, for example, the most stable conformation is one where the hydrogens are staggered relative to each other, while the eclipsed conformation represents an energy maximum. • As the three hydrogens at each end are identical, it is clear that there are three energetically equivalent staggered, and three equivalent eclipsed, conformations. • The rotational energy profile must therefore have three minima and three maxima.
Energi Torsional Etana
The Van der Waals Energy • Evdw is the van der Waals energy describing the repulsion or attraction between atoms that are not directly bonded. • Together with the electrostatic term Eel, it describes the non-bonded energy. • Evdw may be interpreted as the non-polar part of the interaction not related to electrostatic energy due to (atomic) charges. • This may for example be the interaction between two methane molecules, or two methyl groups at different ends of the same molecule.
• Evdw is zero at large interatomic distances and becomes very repulsive for short distances. • In quantum mechanical terms, the latter is due to the overlap of the electron clouds of the two atoms, as the negatively charged electrons repel each other. • At intermediate distances, however, there is a slight attraction between two such electron clouds from induced dipole–dipole interactions, physically due to electron correlation • Even if the molecule (or part of a molecule) has no permanent dipole moment, the motion of the electrons will create a slightly uneven distribution at a given time.
• This dipole moment will induce a charge polarization in the neighbor molecule (or another part of the same molecule), creating an attraction, and it can be derived theoretically that this attraction varies as the inverse sixth power of the distance between the two fragments. • Evdw is very positive at small distances, has a minimum that is slightly negative at a distance corresponding to the two atoms just “touching” each other, and approaches zero as the distance becomes large. • A general functional form that fits these conditions is given in eq. (2. 11).
The Electrostatic Energy: Charges and Dipoles • The other part of the non-bonded interaction is due to internal (re)distribution of the electrons, creating positive and negative parts of the molecule. • A carbonyl group, for example, has a negatively charged oxygen and a positively charged carbon. • At the lowest approximation, this can be modelled by assigning (partial) charges to each atom. • Alternatively, the bond may be assigned a bond dipole moment. These two descriptions give similar (but not identical) results. • Only in the long distance limit of interaction between such molecules do the two descriptions give identical results.
• The interaction between point charges is given by the Coulomb potential, with being a dielectric constant. • The atomic charges can be assigned by empirical rules, but are more commonly assigned by fitting to the electrostatic potential calculated by electronic structure methods
Cross Terms • The first five terms in the general energy expression, eq. (2. 1), are common to all force fields. The last term, Ecross, covers coupling between these fundamental, or diagonal, terms. Consider for example a molecule such as H 2 O. • It has an equilibrium angle of 104. 5° and an O—H distance of 0. 958 Å. If the angle is compressed to say 90 , and the optimal bond length is determined by electronic structure calculations, the equilibrium distance becomes 0. 968Å, i. e. slightly longer. • Similarly, if the angle is widened, the lowest energy bond length becomes shorter than 0. 958Å. This may qualitatively be understood by noting that the hydrogens come closer together if the angle is reduced. • This leads to an increased repulsion between the hydrogens, which can be partly alleviated by making the bonds longer. If only the first five terms in the force field energy are included, this coupling between bond distance and angle cannot be modelled.
• It may be taken into account by including a term that depends on both bond length and angle. Ecross may in general include a whole series of terms that couple two (or more) of the bonded terms. • The components in Ecross are usually written as products of first-order Taylor expansions in the individual coordinates. • The most important of these is the stretch/bend term, which for an A—B—C sequence may be written as in eq. (2. 31)
Aplikasi MM: Padatan Ionik • Aplikasi mekanika molekuler padatan ionik serupa dengan kalkulasi energi kisi • Bahkan metode MM memang bisa digunakan untuk menghitung energi kisi, juga efek adanya cacat pada senyawa ionik dan sifat kristal • Pertanyaan awal untuk mengurai energi kisi: • Apa gaya yang menahan ion-ion berkumpul membentuk kristal pada lattice site –nya • Jawabannya adalah gaya tarik elektrostatik antara muatan positif dan muatan negatif
Aspek Energi dalam Ikatan Ionik: Energi Kisi • Misalkan ada suatu reaksi antara unsur logam yang reaktif (Li) dan mudah melepas elektron dengan gas halogen (F) yang cenderung menarik elektron: Li(g) Li+(g) + e- IE 1 = 520 k. J F(g) + e- F-(g) EA = -328 k. J • Reaksi total: Li(g) + F(g) Li+(g) + F-(g) IE 1 + EA = 192 k. J
• Energi total yang dibutuhkan reaksi ini bahkan lebih besar karena kita harus mengkonversi Li dan F kedalam bentuk gas • Akan tetapi eksperimen menunjukkan enthalpi pembentukan padatan Li. F (∆H 0 f) = -617 k. J • Jika kedua unsur dalam bentuk gas: • Li+(g) + F-(g) Li. F(g) ∆H 0 = -755 k. J • Energi kisi adalah perubahan enthalpi yang menyertai ionion gas yang bergabung membentuk padatan ionik: • Li+(g) + F-(g) Li. F(s) ∆H 0 kisi Li. F = energi kisi = -1050 k. J
Daur Born-Haber
Nilai Energi Born-Haber • Hoatom Li = 161 k. J • BE F 2 = 159 k. J • IE 1 (Li) = 520 k. J • EA (F) = -328 k. J • Ho. Lattice (Li. F) = -1050 k. J • Hof Li. F = -617 k. J • Total Energi : Hof Li. F = Hoatom Li + ½ BE F 2 + IE 1 (Li) + EA (F) + Ho. Lattice
Pendekatan Mekanika Molekuler • Ion-ion diasumsikan berada pada situs kisi masing-masing sesuai dengan muatan formalnya, sehingga Na. Cl misalnya membentuk array of Na+ and Cl- ions. • The net interaction can be obtained by summing the interactions over all the pairs of ions, including not only the attraction between Na+ and Cl- but also the repulsion between ions of the same sign. • The net interaction decreases with distance but slowly so that it is difficult to obtain an accurate value. • To calculate lattice energies, this summation be achieved for simple lattice structures by introducing the Madelung constant. • However, for layer structures with low symmetry this approach is not feasible, as a single Madelung constant will not suffice.
Madelung Constants • There are many factors to be considered such as covalent character and electron-electron interactions in ionic solids. • But for simplicity, let us consider the ionic solids as a collection of positive and negative ions. In this simple view, appropriate number of cations and anions come together to form a solid. • The positive ions experience both attraction and repulsion from ions of opposite charge and ions of the same charge. • The Madelung constant is a property of the crystal structure and depends on the lattice parameters, anion-cation distances, and molecular volume of the crystal
• Before considering a three-dimensional crystal lattice, we shall discuss the calculation of the energetics of a linear chain of ions of alternate signs • Let us select the positive sodium ion in the middle (at x = 0) as a reference and let r 0 be the shortest distance between adjacent ions (the sum of ionic radii). • The Coulomb energy of the other ions in this 1 D lattice on this sodium atom can be decomposed by proximity (or "shells").
• Nearest Neighbors (first shell): This reference sodium ion has two negative chloride ions as its neighbors on either side at r 0 so the Coulombic energy of these interactions is • Next Nearest Neighbors (second shell): Similarly the repulsive energy due to the next two positive sodium ions at a distance of 2 r 0 is
• Next Nearest Neighbors (third shell): The attractive Coulomb energy due to the next two chloride ions neighbors at a distance 3 r 0 is • and so on. Thus the total energy due to all the ions in the linear array is
MM Approach … • Since the computer programs in use are set up to be of general application, they employ methods that give a good approximation to the sum over an infinite lattice for any unit cell. • However, electrostatic interaction is not that has to be considered. We know, for example, that ions are not just point charges but have a size; the shell of electrons around each nucleus prevents too close an approach by other ions. • We therefore include a term to allow for the interaction between shells on the different ions. It would be possible to give each ion a fixed size and insist that the ions cannot be closer than their combined radii. • However, most programs use a different approach by including terms representing intermolecular forces.
• The intermolecular forces act between cations, and between cations and anions, as well as between anions. • For oxides in particular, however, the cation-cation term is often ignored. • Salts such as magnesium oxide can be thought of as close-packed arrays of anions with cations occupying the octahedral holes. • Because the cations are held apart by the anions, the cation-cation interaction is un-important.
Octahedral Hole
• The final thing we need to take into account is the polarizability of the ions. This is a measure of how easily the ions are deformed from their normal spherical shape. • In a perfect crystal, the ions are in very symmetrical environments and can be thought of as spherical. If one ion moves to an interstitial site, leaving its original position vacant, then the environment may not be so symmetrical and it may be deformed by the surrounding ions. • A very simple way to model this is to divide the ionic charge between a core that stays fixed at the position of the ion and a surrounding shell that can move off-center. The distribution of the charge is obtained by adjustment to fit the properties of a crystal containing that ion.
• The shell behaves as though it were attached to the core by springs. Take a chloride ion, for example. If the surrounding ions move so that there is a greater positive charge in one direction, then the shell will move so that the total charge on the ion is distributed over two centers producing a dipole. • Opposing this will be the pull of the springs that attach it to the core.
• For ionic solids, the most important term for lattice energies is the electrostatic term; for sodium chloride, for example, the total lattice energy in a typical calculation is -762. 073 k. J mol-1, of which -861. 135 k. J mol-1 is due to the electrostatic interaction while the intermolecular force and shell terms contribute +99. 062 k. J mol-1. • Thus the contributions of the intermolecular force and shell terms are about 10% of the electrostatic interactions. • These other terms may have a greater relevance in the study of defects.
Crystal Defects in Silver Chloride • Silver halides are used in photography to capture light and form an image. • The action of light on the halide produces silver which forms the black areas of the negative (Figure 2. 2). • The formation of silver depends on the presence of Frenkel defects in the crystal
• Two most common point defects in crystals are Frenkel defects and Schottky defects. What are these? • In Schottky defects, equal numbers of cations and anions are missing (for 1 : 1 structures such as Ag. Cl). • In Frenkel defects, an ion is displaced from its lattice site to an interstitial site; for example, a small cation in a crystal with the Na. Cl structure can move to a tetrahedral hole from the octahedral hole normally occupied. • We can use molecular mechanics to estimate the energies of these defects in silver halides. • The dominant defect in silver halides is a Frenkel defect, in which a silver ion moves to an interstitial site. To calculate the energy required to form this defect we simply remove a silver ion from one position, put it in its new position and compare the energy of the crystal lattice with that of the perfect lattice.
• In a Frenkel defect, there is a vacancy where an ion should be and an ion in a more crowded interstitial position. Would you expect the ions in the vicinity of the defect to stay on their lattice positions? • It would be reasonable to suppose that the ions would adjust their positions to allow the interstitial atom more room, and to take up the space left by the vacancy. • When calculating the energy of formation of the defect the nearest atoms are allowed to adjust their position to obtain the lowest energy for the crystal including the defect. • Figure 2. 3 (overleaf) shows how the chloride ions move when a Frenkel defect forms in Ag. Cl; in the perfect crystal (Figure 2. 3 a) there is just one Ag-Cl bond length, whereas in the defect crystal (Figure 2. 3 b) the Ag-C 1 bond lengths are shortened and variable.
(a) Perfect Ag. Cl. (b) A Frenkel defect in Ag. Cl (a) (b) • For an estimate of the actual numbers of defects we need to know the Gibbs energy of formation, but the major contribution comes from the internal energy. • Calculated values for the energy of formation of cation Frenkel defects in Na. Cl and Ag. Cl are 308 k. J mol-1 and 154 k. J mol-l, respectively.
Zeolite • Zeolites* have frameworks of silicon, aluminium and oxygen atoms which form channels and cages, e. g. Figure 2. 4. • They form a wide variety of structures but all are based on silicon tetrahedrally bound to oxygen. • Differing numbers of silicon atoms are replaced by aluminium. Other cations, notably those of Groups I, II and the lanthanides, are present in the structures to balance the charge. • Surprisingly such structures can be very successfully modelled by considering them as a collection of ions and using the methods discussed
• Silicon is not normally thought of as forming Si 4+ ions; indeed silica, SO 2, and silicates do contain silicon covalently bonded to oxygen • We do have to make some allowance for the covalency of the Si-0 bonds. • The most successful way of doing this is to add a term that represents the resistance of OSi. O and OAl. O bond angles to deviation from the tetrahedral angle. • The covalency of zeolites and related compounds is also reflected in the relative size of the electrostatic and other terms
• For one form of silica, SO 2, for example, a calculated lattice energy of -12416. 977 k. J mol-1 had contributions of -16029. 976 k. J mol-1 from electrostatic interactions, +3553. 796 k. J mol-1 from intermolecular force terms and the core-shell spring term, and 1. 913 k. J mol-1 from those OSi. O bond angles that were not tetrahedral. • Here the intermolecular force terms are about 20% of the electrostatic interaction. • The energy due to the term keeping the angles tetrahedral is small, but without this term the zeolite structure is lost. • With this addition, the structures of a wide variety of zeolites, both naturally occurring minerals and synthetic zeolites tailored to act as catalysts, can be modelled and then used to answer questions such as which position will the non-framework ions and molecules occupy and how do ions travel through the structure?
Modelling Organic Molecules • The power of this method for organic molecules lies in the adoption of a relatively small set of parameters that can be transferred to any molecule you want. • But what sort of parameters might be needed? Can we simply use electrostatic and intermolecular forces? How do we allow for bonds and different conformations • Let us start by looking at a very simple molecule - ethane. Ethane is H 3 C-CH 3. • As for solids, we do need to include an electrostatic interaction, but what charge are we going to give carbon and hydrogen atoms? • Obviously +4 or -4 on C and +I or -1 on H are unrealistic and would not even give a neutral molecule. • Think for a moment about the process of bond formation
• When two atoms form a covalent bond, they share electrons. If the atoms are unalike then one atom has a larger share than the other, resulting in a positive charge on one atom and a negative charge on the other. • But the charge transferred is less than one electron. For diatomic molecules, the charge on each atom can be obtained experimentally. • In the molecule HCI, for example, the hydrogen atom has a charge of +O. 18 and the chlorine atom a charge of -0. 18. • The fractional charges are known as partial charges
• A convenient way of setting up a set of transferable partial charges is to give each atom a contribution to the partial charge from each type of bond that it is involved in. • For example, in chloroethane, CH 3 CH 2 Cl, we need to consider contributions for the carbon atoms for carbon bound to carbon, carbon bound to hydrogen and carbon bound to chlorine. • Carbon bound to carbon is given a value of zero. • For elements such as oxygen, which can be singly or doubly bonded (C-O or C=O), we need different partial charge contributions for each type of bond • In one available computer program carbon bonded to hydrogen gives a contribution of +0. 053 • As well as the electrostatic interaction arising from the partial charges, we also need intermolecular forces. • These can be important for large atoms such as bromine or iodine
Cocaine analogue - Ecgonine • List all the bond terms you would need to describe the compound on the left