Mediterranean Anemia Thalassemia Kakavoulis Nikolaos Patras Ioannis What







 • asymptomatic • microcytosis • minor")





 Regular")


- Slides: 21
Mediterranean Anemia. Thalassemia Kakavoulis Nikolaos Patras Ioannis
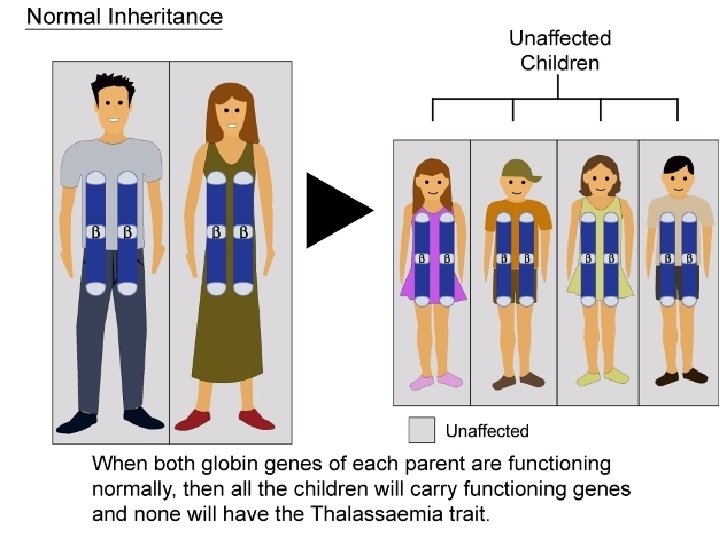
What is Thalassaemia ? Thalassaemia is a group of inherited disorders of hemoglobin synthesis characterized by reduced or absence of one or more of the globin chains of adult hemoglobin. Genetically, it is autosomal recessive blood disease. The name is derived from the Greek words Θάλασσα= Sea" and ”Αίμια= Blood" in reference to anemia of the sea.
Demographics: Thalassemia • Found most frequently in the Mediterranean, Africa, Western and Southeast Asia, India and Burma • 15% of the greek population have the ‘’T’’ gene.
Genetic. Types of Thalassaemia : There are two basic groups of thalassaemia. q Alpha ( )Thalassaemia q Beta ( )Thalassaemia
Normal Human Haemoglobins Haemoglobin Structural formula Adult Hb-A 2 2 2 97% 2 2 1. 5 -3. 2% Fetal (And 1% in adults) Embryonic Hb-F 2 2 Hb-Gower 1 Hb-Gower 2 Hb-Portland 2 2 2 2 0. 5 -1%
Chromosomes
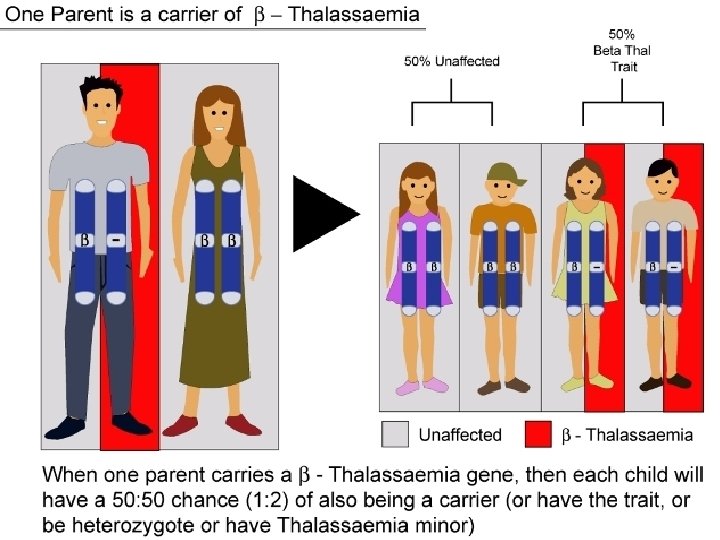
β Thalassemia β Thalassemia: deficient/absent beta subunits Commonly found in Mediterranean, Middle East, Asia, and Africa Three types: Minor Intermedia Major (Cooley anemia) May be asymptomatic at birth as Hb. F functions
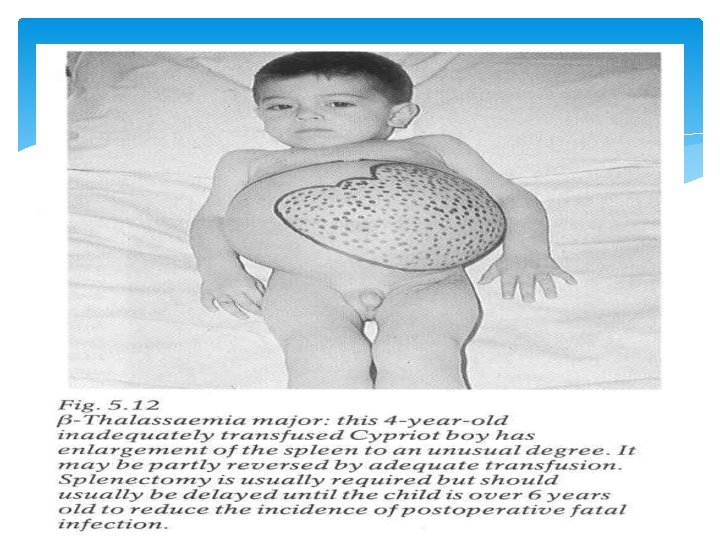
Clinical Outcomes of β-Thalassemia β Thalassemia minor (trait) • asymptomatic • microcytosis • minor anemia β Thalassemia intermedia • symptoms similar to Cooley Anemia but less severe β Thalassemia major (Cooley Anemia) • • most severe form moderate to severe anemia intramedullary hemolysis (RBC die before full development) peripheral hemolysis & splenomegaly skeletal abnormalities (overcompensation by bone marrow) increased risk of thromboses pulmonary hypertension & heart failure
Pathophysiology Disturbance of ratio between α & non-α globin chain synthesis then absence or decrease production of one or more globin chains Formation of abnormal Hb structures Ineffective erythropoiesis Excessive RBCs Destruction Iron Overload Extra-medullary hematopoiesis Increased Hb. F expression
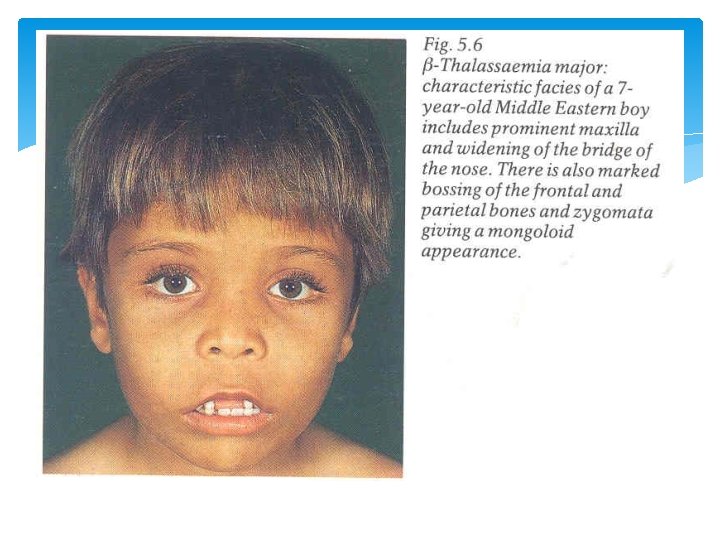
Signs & Symptoms Thalassaemia Minor : Usually no signs or symptoms except for a mild anemia. Thalassaemia Major : 1. Paleness, Jaundice or yellow coloured skin. 2. Growth retardation. 3. Bony abnormalities specially of the facial bones. 4. Enlarged spleen and liver.
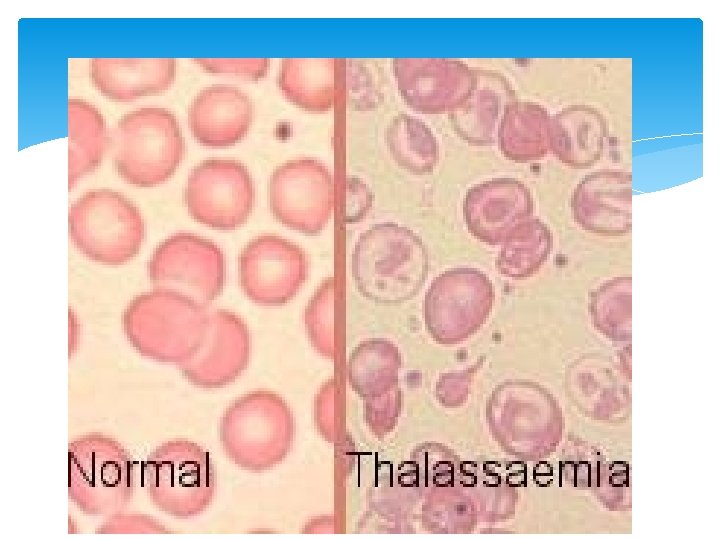
Laboratory Diagnosis Thalassemia minor: ØHaemoglobin : Haemoglobin level is usually normal or mildly reduced. ØPeripheral blood film : Hypochromia and Microcytosis (similar to Iron Deficiency Anemia). ØMCV< 75 fl, RDW < 14%. ØReticulocyte Count increases ØDecrease Osmotic Fragility ØHaemoglobin electrophoresis
Other Special Procedures Globin Chain Testing - determines ratio of globin chains being produced. DNA Analysis - Determine specific defect at molecular DNA level. 17
Course and treatment of thalassaemia If Untreated v thalassemia Major : Death in first or second decade of life v. Intermedia: variable life span v. Minor/Minima: Normal life span
Treatment for β Thalassemia Trait – no treatment required Intermedia Major (Cooley anemia) Regular folate supplementation RBC transfusion (Splenectomy may decrease need for transfusions) to maintain [Hgb] ~9 -10 g/d. L Blood transfusions iron accumulation iron overload Iron chelators (diferroxamin)
Suggestions for encountering the disease in a more efficient way Raising awareness for more frequent blood donations, since patients with β-thalasseamia require frequent transfusions 8 th of May: Thalassemia awareness day.
References http: //www. mayoclinic. com/health/thalassemia/DS 00 905/DSECTION=treatments%2 Dand%2 Ddrugs http: //www. nlm. nih. gov/medlineplus/ency/article/000 587. htm http: //www. lpch. org/Disease. Health. Info/Health. Library /hematology/thalbeta. html http: //www. nhlbi. nih. gov/healthtopics/thalassemia/