MALATTIE NEUROLOGICHE RARE NUOVE PROSPETTIVE TERAPEUTICHE Malattie neuromuscolari













 amyloidosis (h. ATTR) ü Hereditary transthyretin (TTR) amyloidosis (h. ATTR) is")

 modified ASO")

: 419 -25")



- Slides: 23
MALATTIE NEUROLOGICHE RARE: NUOVE PROSPETTIVE TERAPEUTICHE Malattie neuromuscolari Michelangelo Maestri Tassoni
Storia naturale Distrofia Muscolare di Duchenne Precoci difficoltà deambulatorie, frequenti cadute, andatura anserina Precoce perdita della deambulazione (12 anni) Progressivo e generalizzato difetto di forza, conservata la motilità distale delle dita. Sebbene il coinvolgimento della muscolatura sia generalizzato, alcuni gruppi muscolari sono più interessati di altri e questo può determinare coinvolgimenti asimmetrici e favorire lo sviluppo di retrazioni articolari e scoliosi. III decade Muscoli respiratori Insufficienza respiratoria Miocardio cardiomiopatia dilatativa e Scompenso cardiaco Muscoli faringei aspirazione e disfagia Alterazione della muscolatura del tratto gastrointestinale Biopsia muscolare nella DM di Duchenne
Lancet Neurology 2010, 2018
Distrofia muscolare di Duchenne Terapia farmacologica Terapia genica Deflazacort 0, 9 mg/kg/die Prednisone 0, 75 mg/kg/die Riduzione infiammazione Inibitore Miostatina Promotore Urotrofina Idebenone Rigenerazione muscolare Distrofina alternativa Antiossidante Miglioramento funzionalità Respiratoria Inibitore Istone deacetilasi Riduzione infiammazione/fibrosi Rigenerazione muscolare Exon Skipin 45/51/53 Readtrhrough ribosomiale Vettori virali
Translarna è indicato per il trattamento della distrofia muscolare di Duchenne conseguente a una mutazione nonsense nel gene della distrofina (nonsense mutation Duchenne Muscular Dystrophy, nm. DMD) nei pazienti deambulanti di età pari ad almeno 5 anni. L’efficacia non è stata dimostrata nei pazienti non deambulanti.
TERAPIA GENICA NELLA DISTROFIA MUSCOLARE DI DUCHENNE Eteplirsen, commercially available as Exondys 51™ in the USA, obtained in September 2016 a conditional approval by FDA to treat DMD patients amenable to exon 51 skipping (13% of DMD patients). IV administration
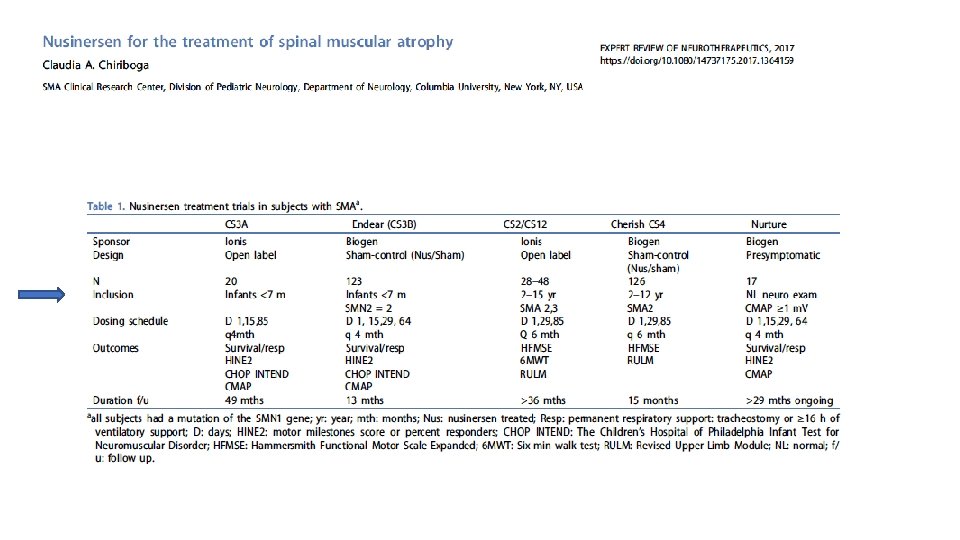
AMIOTROFIA SPINALE SMA 1: esordio 0 -6 mesi SMA 2: esordio 6 -18 mesi SMA 3: esordio >12 mesi SMA 4: esordio in età adulta 1/10000 nati vivi La più comune causa genetica di morte infantile Due copie altamente omologhe del gene
AMIOTROFIA SPINALE Autosomal recessive inherited disorder usually caused by a deletion or mutation in the survival motor neuron 1 (SMN 1) gene on chromosome 5 q. The mutation is most commonly a homozygous deletion involving exon 7 of the gene In humans, the SMN protein is encoded by the SMN 1 and SMN 2 genes. The C to T substitution in exon 7 of SMN 2 is translationally silent, but alters splicing such that the majority of SMN 2 transcripts lack exon 7 and the truncated protein is unstable. Normally, SMN 1 produces abundant SMN protein. In SMA, homozygous mutation of SMN 1 results in only a small amount of functional SMN protein contributed by the varying copy numbers of SMN 2
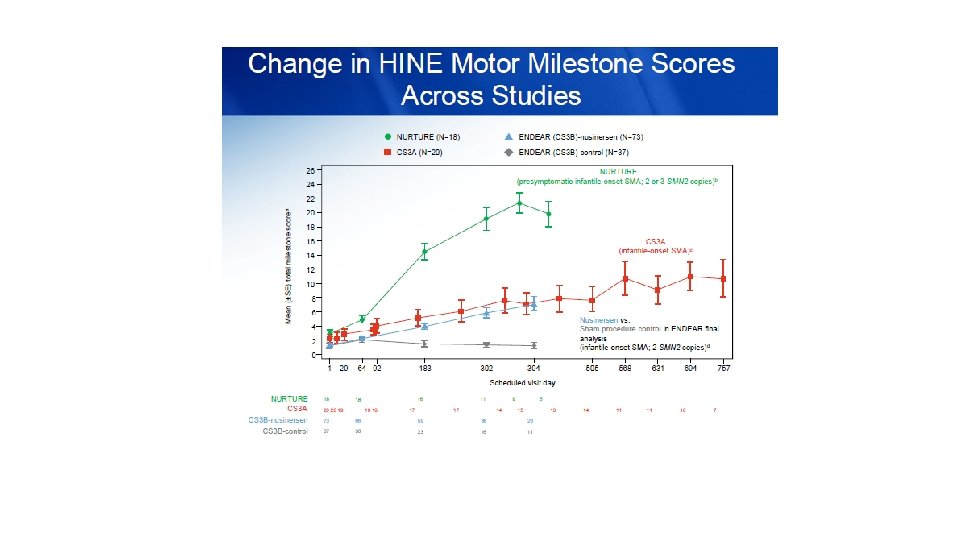
Oligonucleotide antisenso che inattiva un sito di splicing a livello di SMN 2, aumentando la stabilità dei trascritti SMN 2 e i livelli di proteine SMN NUSINERSEN Dicembre 2016!
NEL DICEMBRE 2016 IL NUSINERSEN E’ DIVENTATO IL PRIMO FARMACO APPROVATO DALLA FDA PER LA SMA
Administration as intrathecal bolus injection over 1 to 3 minutes using a spinal anesthesia needle Osservazione in ambiente ospedaliero per 24 ore
Risdiplam ü an orally administered, centrally and peripherally distributed, small-molecule SMN 2 m. RNA splicing modifier ü It promotes similar increases of SMN protein levels in the brain and peripheral organs of SMA mice ü After demonstration of safety and tolerability in a phase 1 study in male volunteers with escalating dose (Br J Pharmacol, 2019), still ongoing phase 2 and 3 clinical trials started in SMA 1, 2, and 3 patients
Hereditary transthyretin (TTR) amyloidosis (h. ATTR) ü Hereditary transthyretin (TTR) amyloidosis (h. ATTR) is a progressive devastating disease transmitted as an autosomal dominant trait, characterized by multiple organ failure including axonal sensory-motor neuropathy, cardiac involvement and autonomic dysfunction, kidney and gastrointestinal involvement and vitreous opacities ü h. ATTR onset occurs between the third and eighth decades of life and patients are classified having early (before 35 years) or later (after 55 years) onset Tafamidis, inotersen and patisiran
Tafamidis: risposta neurologica nel 50% circa dei pazienti, riduzione della mortalità cardiovascolare Cortese et al. , 2016 Maurer et al. , 2018
Inotersen inhibits hepatic TTR production. It is a second generation 2′-O-(2 -methoxyethyl) modified ASO which is complimentary to a region in the 3′ untranslated region of the human wild-type and all known amyloidogenic variant TTR m. RNA. This hybridization leads to an RNase H 1 -mediated degradation of TTR m. RNA, thus preventing TTR production Patisiran is the first-ever small interfering RNA (si. RNA) therapeutic. It is encapsulated in a lipid nanoparticle delivered to intracellular compartments of hepatocytes. It specifically binds to the 3′ untranslated region of mutant and wild-type TTR m. RNA, reducing both mutant and wild-type TTR production Subcutaneous administration Intravenous administration; infusion reaction
EVADARONE NELLA SLA Criteri di inclusione AIFA: diagnosi di SLA definita o probabile secondo i criteri rivisti di El Escorial; età ≥ 18 anni; punteggio ≥ 2 in ogni item della scala validata come strumento di monitoraggio della progressione di disabilità (ALSFRS-r); capacità vitale forzata (CVF) ≥ 80% del teorico; evidenza di progressione di malattia negli ultimi 3 mesi. Lo schema di trattamento prevede la somministrazione di Edaravone 60 mg (2 flaconi da 30 mg/100 ml) endovena in 60 minuti a cicli di 28 giorni: nel ciclo iniziale Edaravone viene somministrato per 14 giorni consecutivi seguiti da 2 settimane di pausa; dal secondo ciclo in poi la somministrazione avviene per 5 giorni consecutivi/settimana per 2 settimane, seguiti da due settimane di pausa.
Sanders DB et al. Neurology 2016 Jul 26; 87(4): 419 -25
The schedule for eculizumab was induction dosing 900 mg on day 1 and weeks 1, 2, and 3; 1200 mg at week 4; and maintenance dosing 1200 mg every second week thereafter.
Nuovi approcci terapeutici nella miastenia gravis
Living the era of a therapeutic revolution üNecessità di diagnosi precoce üSfide organizzative (terapia endovenose con schemi complessi, terapie intrarachidee) üCosti üAccessiblità üCriteri di inclusione ed esclusione üMonitoraggio effetti collaterali üAIFA/pazienti