LIPID STORAGE DISEASES SPHINGOLIPIDS Sphingolipids are important constituents






 are a group of inherited diseases that are often")
 Defects give rise to swollen lysosomes, developmental and degenerative")

 Autosomal recessive diseases Individually rare Collectively occur @ 1/8000")






 First described in 1880’s from ‘cherry red’ spot in fundus")



. The incidence is particularly")
. This is a severe form of Tay-Sachs disease. Onset usually")







")
,")
 causes little to no damage")










: Disease $) Treatment")



- Slides: 54
LIPID STORAGE DISEASES
SPHINGOLIPIDS Sphingolipids are important constituents of cell membranes. Although sphingolipids contain no glycerol, they are similar in structure to the glycerophos pholipids in that they have a hydrophilic region and two fatty acid derived hydrophobic tails.
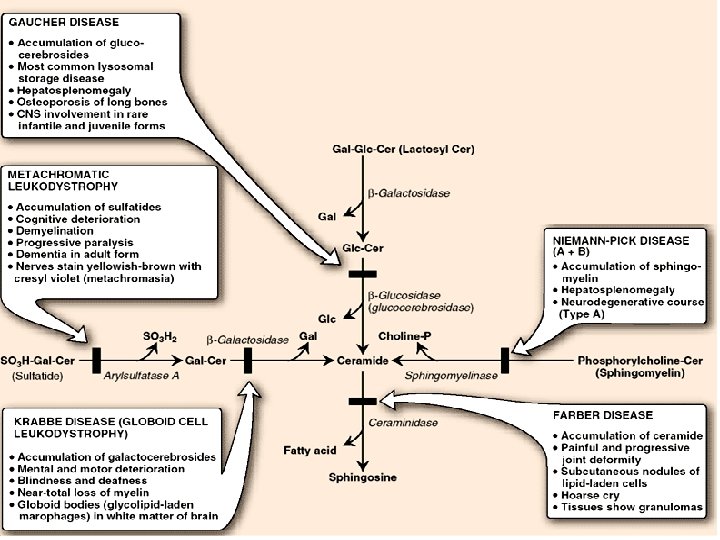
Degradation of glycosphingolipids Glycosphingolipids are internalized by endocytosis. All of the enzymes required for the degradative process are present in lysosomes, which fuse with the endocytotic vesicles. The lysosomal enzymes hydrolytically and irreversibly cleave specific bonds in the glycosphingolipid. Degradation is a sequential process following the rule “last on, first off, ” in which the last group added during synthesis is the first group removed in degradation. [Note: Defects in the degradation of the polysaccharide chains in these three glycoconjugates also result in lysosomal storage diseases. ]
Genetic Deficiencies Catabolism of Enzymes in Sphingolipids released when membrane is degraded are digested in endosomes after fusion with lysosomes. Lysosomes contain many enzymes, each of which removes specific groups from individual sphingolipids. Genetic deficiencies of many of these enzymes are known, and the diseases share some of the characteristics.
Certain diseases are characterized by abnormal quantities of these lipids in the tissues, often in the nervous system. They may be classified into two groups: (1) true demyelinating diseases and (2) sphingolipidoses. In multiple sclerosis, which is a demyelinating disease, there is loss of both phospholipids (particularly ethanolamine plasmalogen) and of sphingolipids from white matter. Thus, the lipid composition of white matter resembles that of gray matter.
INBORN ERRORS OF PHOSPHOLIPID, SPHINGOLIPID METABOLISM • Sphingolipidosis: • • Accumulation of complex lipids Synthesis of complex lipids is not effected Lack of specific (hydrolytic) lysomal enzymes All tissues are effected • Demyelination: Multiple Sclerosis • White matter decrease of [phospholipid], [plasmenyl ethanolamine], [sphingolipid] Biosynthesis of membrane lipids and steroids 1 7
The sphingolipidoses (lipid storage diseases) are a group of inherited diseases that are often manifested in childhood. These diseases exhibit several constant features: (1) Complex lipids containing ceramide accumulate in cells, particularly neurons, causing neurodegeneration and shortening the life span. (2) The rate of synthesis of the stored lipid is normal. (3 The enzymatic defect is in the lysosomal degradation pathway of sphingolipids. (4) The extent to which the activity of the affected enzyme is decreased is similar in all tissues.
The sphingolipidoses (lipid storage diseases) Defects give rise to swollen lysosomes, developmental and degenerative defects with varying involvement of the nervous system due to ‘storage’ of material in the lysosome. LSDs cause death in childhood (generally) after normal infancy. LSDs are essentially incurable, but some are treatable to varying degrees.
LYSOSOMAL STORAGE DISEASE (Amaurotic Idiocy) Autosomal recessive diseases Individually rare Collectively occur @ 1/8000 live births Cause death in early to late childhood (after normal infancy) Varying involvement of the nervous system All ‘store’ material in the lysosome due to defects in substrate degradation or biogenesis of the lysosome
GENERAL OUTLINE OF LSD DYSFUNCTION: Mutations arising in hydrolytic enzyme, co factor or factor essential of enzyme delivery to lysosome Also, factors essential for lysosome function and biogenesis (membrane proteins, channels and proteins of unknown function) plus factors for protein traffic to lysosome Material (substrate) continues to be delivered to lysosome resulting in ‘stored’ material, leads to swollen lysosomes Developmental dysfunction and early death: symptoms v. variable, varying involvement of different tissues
Usually, lack of an enzyme leads to accumulation of a particular lipid, causing symptoms such as enlarged spleen and liver, seizures, blindness, mental retardation and death Synthesis of complex lipids is not effected • All tissues are effected
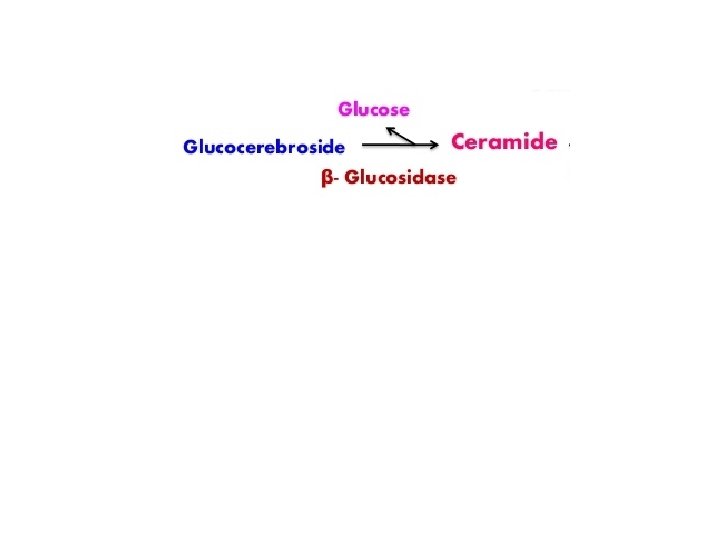
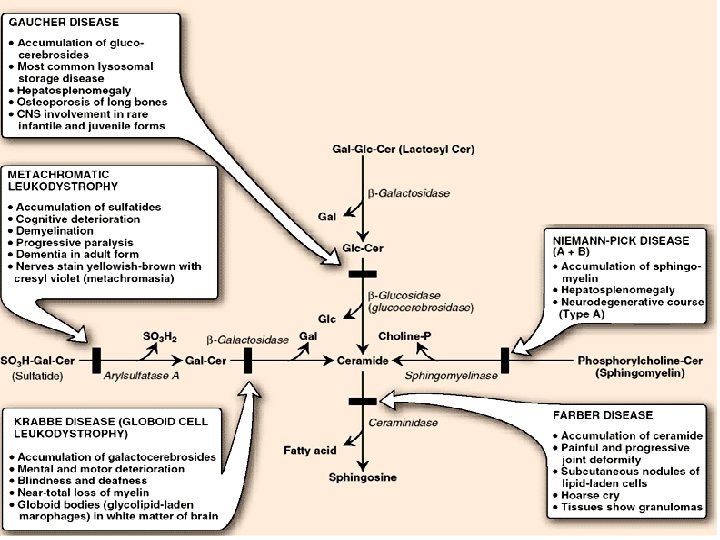
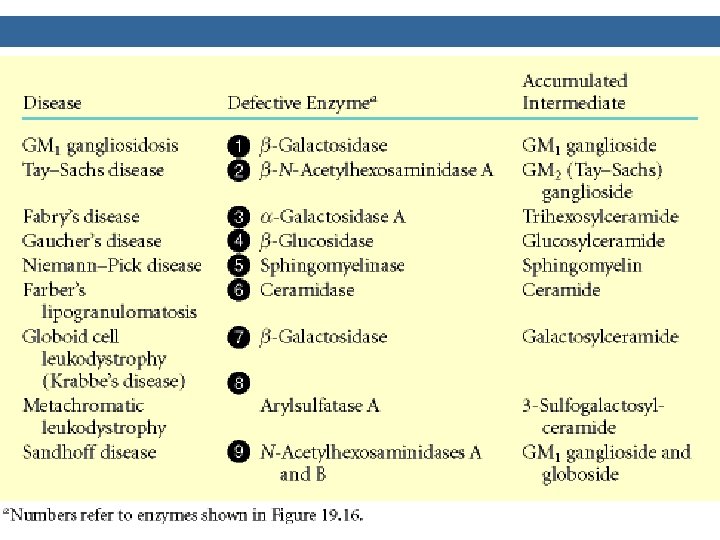
Degradation of sphingolipids Ø by lysosomal enzymes (deficiencies result in lysosomal storage disease = sphingolipidoses) SPHINGOLIPIDOSES genetic mutations, mental retardation, death Name Deficient enzyme Acccumlated lipid Fucosidosis α-Fucosidase H-Isoantigen Generalized gangliosidosis GM 1 -β-Galactosidase GM 1 -Ganglioside Tay-Sachs disease Hexosaminidase A GM 2 -Ganglioside Tay-Sachs variant Hexosaminid. A and B GM 2 -Ganglioside Fabry disease α-Galactosidase Globotriaosylceramide Ceramide lactoside lipidosis Ceramide lactosidase Ceramide lactoside Metachromatic leukodystrophy Arylsulfatase A 3 -Sulfogalactosylceramide Krabbe disease β-Galactosidase Galactosylceramide Gaucher disease β-Glucosidase Glucosylceramide Niemann-Pick disease Sphingomyelin Farber disease Ceramide
GANGLIOSIDOSIS A disease of the accumulation of gangliosides is called "Gangliosidosis", which is a form of Lipid storage disorder. The gangliosidoses are two distinct genetic groups of diseases. Both are autosomal recessive and affect males and females equally. • GM 1 gangliosidoses • GM 2 gangliosidoses
GANGLIOSIDOSIS • The GM 1 gangliosidoses are caused by a deficiency of beta -galactosidase, with resulting abnormal storage of acidic lipid materials in cells of the central and peripheral nervous systems, but particularly in the nerve cells. • GM 1 has three forms: early infantile, late infantile, and adult.
THE GM 2 GANGLIOSIDOSES cause the body to store excess acidic fatty materials in tissues and cells, most notably in nerve cells. these disorders result from a deficiency of the enzyme beta hexosaminidase. the gm 2 disorders include:
Tay-Sachs (GM 2 -gangliosidosis) First described in 1880’s from ‘cherry red’ spot in fundus (retina) (lipid deposition in bipolar ganglion cells) Genetic defect: Hexosaminidase A (HEXA) storage material: GM 2 ganglioside, globoside, glycolipids
Cherry red macular spot in an infantile patient with GM 2 gangliosidosis variant B (Tay–Sachs disease). The dark red spot in the middle of the light central area is secondary to lipid storage in neuronal cells in this area; the storing cells have lost their processes that normally cover the fovea centralis. The fovea normally appears as yellow, but is changed to the red macular spot showing the color of the choroidea behind the retina.
Tay – Sachs Disease Deficiency of Hexosaminidase A hydrolyses the N acetyl galactosamine of GM. 2 ganglioside Ceramid Glu Gal. NAc Gal NANA hexosaminidase Biosynthesis of membrane lipids and steroids 1 21
Tay – Sachs Disease • • Mental retardation Macrocephalia Death at age 2 4 Frequency: Ashkenazi Jewish population 1: 3500 Biosynthesis of membrane lipids and steroids 1 22
Tay Sachs disease (also known as GM 2 variant B). The incidence is particularly high among Eastern European and Ashkenazi Jewish populations, as well as certain French Canadians and Louisianan Cousins. Affected children appear to develop normally for the first few months of life. Symptoms begin by 6 months of age and include progressive loss of mental ability, dementia, decreased eye contact, increased startle reflex to noise, progressive loss of hearing leading to deafness, difficulty in swallowing, blindness, cherry-red spots in the retinas, and some paralysis. Seizures may begin in the child’s second year. Children may eventually need a feeding tube and they often die by age 4 from recurring infection. NO SPECIFIC TREATMENT IS AVAILABLE. Anticonvulsant medications may initially control seizures. Other supportive treatment includes proper nutrition and hydration and techniques to keep the airway open. A much rarer form of the disorder, which occurs in patients in their twenties and early thirties, is characterized by unsteadiness of gait and progressive neurological deterioration.
SANDHOFF DISEASE (VARIANT AB). This is a severe form of Tay-Sachs disease. Onset usually occurs at the age of 6 months and is not limited to any ethnic group. Neurological symptoms may include progressive deterioration of the central nervous system, motor weakness, early blindness, marked startle response to sound, spasticity, myoclonus (shock-like contractions of a muscle), seizures, macrocephaly (an abnormally enlarged head), and cherry-red spots in the eye. Other symptoms may include frequent respiratory infections, murmurs of the heart, doll-like facial features, and an enlarged liver and spleen. There is no specific treatment for Sandhoff disease. As with Tay-Sachs disease, supportive treatment includes keeping the airway open and proper nutrition and hydration. Anticonvulsant medications may initially control seizures. Children generally die by age 3 from respiratory infections.
Current Research • Enzyme replacement therapy to provide the Hex-A which is missing in babies with Tay. Sachs disease has not been successful due to the blood-brain barrier, which prevents Hex. A from entering the brain through the blood • Bone marrow transplantation has also been attempted but to date has not been successful in reversing or slowing damage to the central nervous system in babies with Tay-Sachs disease
A, Near end stage in a Spanish 13 year old patient with GM 2 gangliosidosis variant B 1 (photograph courtesy of H. H. Goebel, Mainz ).
B, GM 2 gangliosidosis in a 23 week old fetus; and part of a cortical neuron; early stage of membranous cytoplasmic bodies (two clusters marked by crosses), i. e. , lipid storing secondary lysosomes, × 36, 000 (courtesy W. Schlote, Tübingen). • .
C, The large cells are neurons distended by lysosomal storage in the medulla oblongata of a 1. 5 year old patient with GM 1 gangliosidosis. The small dark bodies are nuclei from increased numbers of glial cells (“reactive gliosis”). • .
Degradation of Sphingomyelin • Sphingomyelin is degraded by sphingomyelinase, a lysosomal enzyme that hydrolytically removes phosphorylcholine, leaving a ceramide. • The ceramide is, in turn, cleaved by ceramidase into sphingosine and a free fatty acid
Sphingolipidoses • . Ceramide Niemann-Pick Disease P-Choline sphingomyelinase • Accumulation of sphingomyelin Biosynthesis of membrane lipids and steroids 1 30
SPHINGOMYELIN DEGRADATION • Niemann Pick disease is due to sphingomyelinase deficiency • Sphingomyelin accumulates in the brain, spleen, and liver
Types of Niemann Pick disease • • Niemann Pick disease (Types A and B) Autosomal recessive disease Inability to degrade sphingomyelin The deficient enzyme is sphingomyelinase—a type of phospholipase C
Niemann Pick disease. In the severe infantile form (Type A—less than 1% normal activity), the liver and spleen are the primary sites of lipid deposits and are, therefore, tremendously enlarged. The lipid consists primarily of the sphingomyelin that cannot be degraded. Infants with this lysosomal storage disease experience rapid and progressive neurodegeneration as a result of deposition of sphingomyelin in the central nervous system, and they die in early childhood
A less severe variant (Type B— 5% or more) causes little to no damage to neural tissue, but lungs, spleen, liver, and bone marrow are affected, resulting in a chronic form of the disease, with a life expectancy into adulthood. Although Niemann Pick disease occurs in all ethnic groups, Type A occurs with greater frequency in the Ashkenazi Jewish population than in the general population.
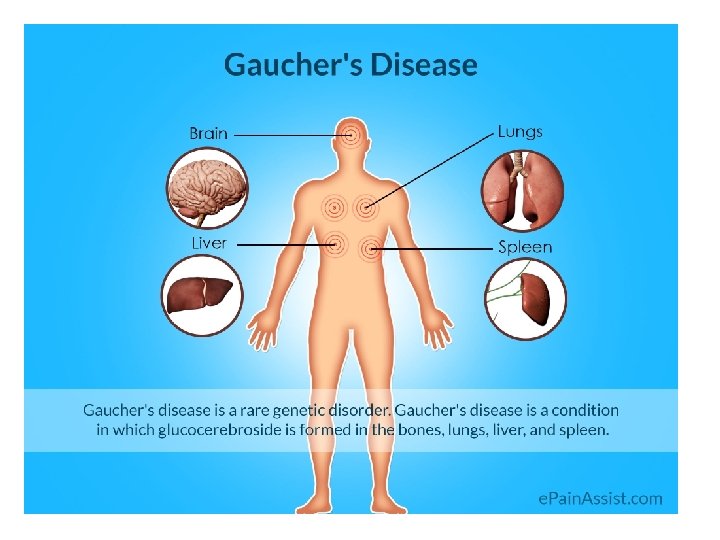
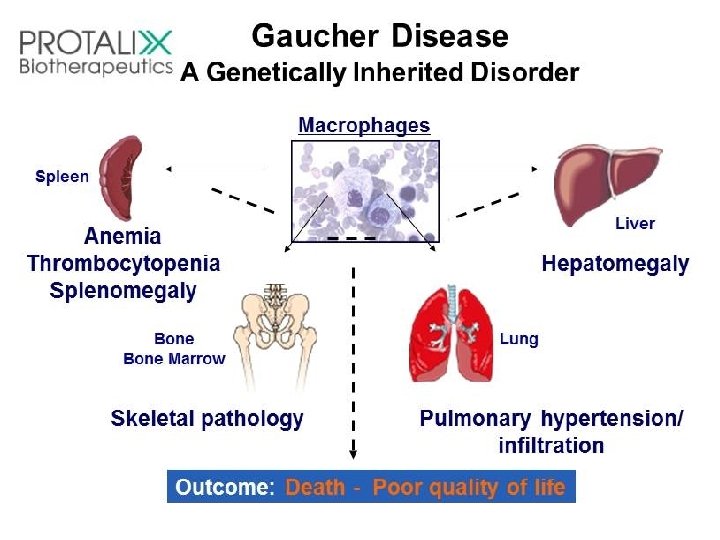
Gaucher’s - disease • Accumulation of glucocerebrosides in reticuloendothelial cells of spleen, liver, and bone marrow • the most common of lysosomal storage disease • Mental retardation • . Biosynthesis of membrane lipids and steroids 1 35
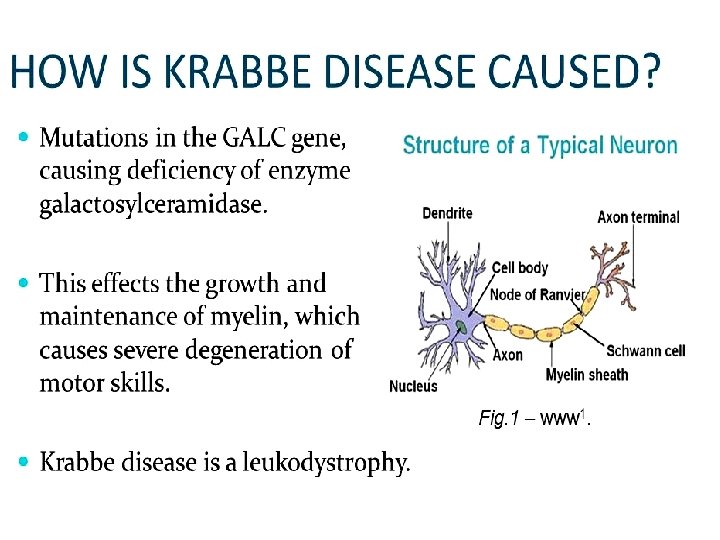
Krabbe disease • Accumulation of Galactosyl ceramide Ceramide Gal • . b-galactosidase Biosynthesis of membrane lipids and steroids 1 39
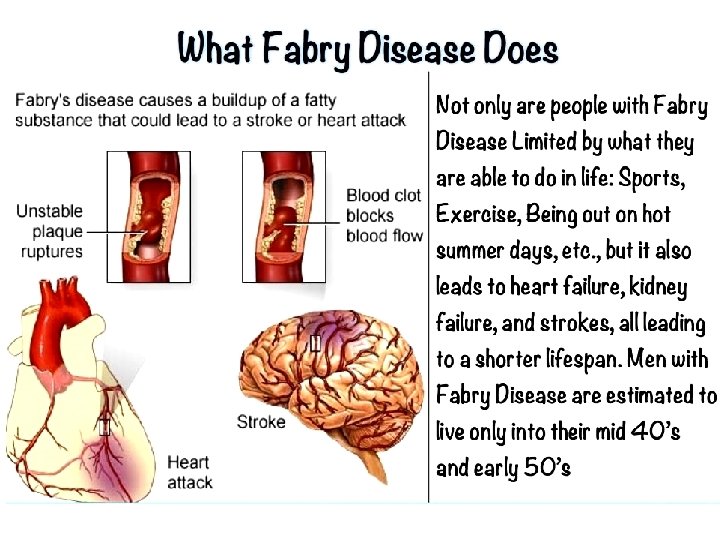
Sphingolipidoses Ceramide Glu Fabry Disease Gal a-galactosidase Biosynthesis of membrane lipids and steroids 1 44
SUMMARY OF DEFECTS IN GANGLIOSIDE CATABOLISM THAT CONTRIBUTE TO HUMAN DISEASE Medical Condition Defective Enzyme Major Accumulating Metabolite Generalized Gangliosidosis Beta-Galactosidase GM 1 Tay-Sachs Disease Hexosaminidase A GM 2 Gaucher’s Disease Glucocerebrosidase Glucosylcerebroside Niemann-Pick Disease Sphingomyelin Sandhoff’s Disease Hexosaminidase A&B A Globoside* Fabry’s Disease Alpha-Galactosidase A A Globoside* *The globosides that are substrates for the defective enzymes in these diseases do not have specific names, but are referred to by the broad term globoside.
ECONOMIC COST ERT is current most effective treatment (non neurodegenerative LSDs): Disease $) Treatment Annual Cost (per patient in Gaucher ERT 145, 000 290, 000 Gaucher SRT 91, 000 Fabry ERT 156, 000 Reasons: High regulatory costs Cost of research Lack of competition (Orphan Drug Act 1983, US)
Some sphingolipidoses
Diagnosis and treatment: A specific sphingolipidosis can be diagnosed by measuring enzyme activity in cultured fibroblasts or peripheral leukocytes, or by analysis of DNA (see p. 473). Histologic examination of the affected tissue is also useful. [Note: Shell like inclusion bodies are seen in Tay Sachs, and a wrinkled tissue paper appearance of the cytosol is seen in Gaucher disease (Figure 17. 19). ] Prenatal diagnosis, using cultured amniocytes or chorionic villi, is available. Gaucher disease, in which macrophages become engorged with glucocerebroside, and Fabry disease, in which globosides accumulate in the vascular endothelial lysosomes of the brain, heart, kidneys, and skin, are treated by recombinant human enzyme replace ment therapy, but the mone tary cost is extremely high. Gaucher has also been treated by bone marrow transplantation (because macrophages are derived from hematopoietic stem cells), and by substrate reduction therapy through pharmacological reduction of glucosylceramide, the substrate for the deficient enzyme.
There is no effective treatment for many of the diseases, though some success has been achieved with enzymes that have been chemically modified to ensure binding to receptors of target cells, eg, to macrophages in the liver in order to deliver β glucosidase (glucocerebrosiase) in the treatment of Gaucher’s disease. A recent promising approach is substrate reduction therapy to inhibit the synthesis of sphingolipids, and gene therapy for lysosomal disorders is currently under investigation.