LEUKEMIA DR SUBHAN ALI R PATHOLOGY LEUKEMIA INTRODUCTION

LEUKEMIA DR SUBHAN ALI R PATHOLOGY

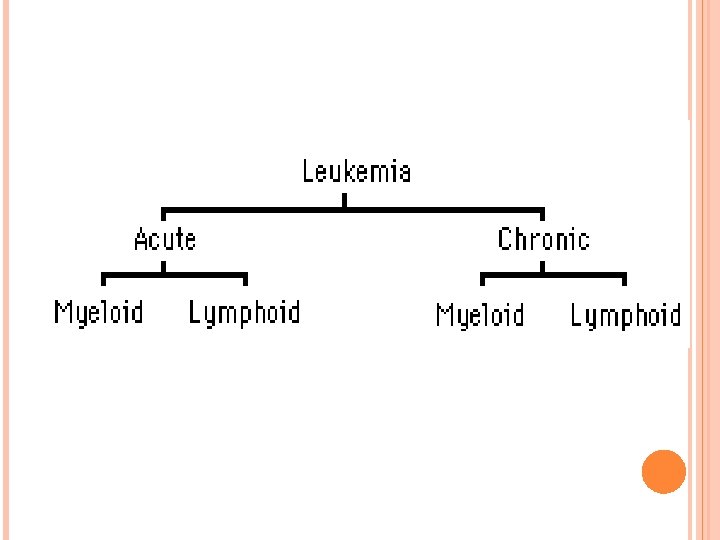
LEUKEMIA : INTRODUCTION Leukemia is a malignant neoplasm of hematopoietic tissue originating in and infiltrating the bone marrow. Leukemia generally involves the peripheral blood, and often infiltrates the spleen, liver, and lymph nodes

INCIDENCE Approximately 10% of all malignant neoplasms about 27, 800 new cases/year and 18, 100 deaths/year (1990). The incidence rate is almost 10 new cases /100, 000 people in the US. The number of new cases is split evenly between acute and chronic leukemia. Of the 27, 800 new cases, 2, 500 are in children (<15 years). Leukemia occurring in children is almost always acute with a 4: 1 ratio of lymphoid to myeloid leukemia. This is the reverse of that seen in adult leukemia.
 adult leukemia and 45% of all")
INCIDENCE AML is the most common (80 -90%) adult leukemia and 45% of all leukemias. ALL peaks at age 4, - 10% of all leukemias. CLL is rare before the age of 40 and almost never seen in childhood, 30% of all leukemias. CML peaks at age 30 -50, but can occur at any age, 15% of all leukemias

EPIDEMOLOGY There is approximately a 20 fold increase incidence of leukemia in children with Down's syndrome. Other disorders (autosomal recessive) associated with leukemia include Bloom's syndrome, Fanconi's anemia, and ataxia telangiectasia (all disorders with defective DNA excision &/or repair). Immunodeficiency diseases (ataxia telangiectasia; agammaglobulinemia) may also be associated with leukemia There is no apparent predilection for race or sex among the myeloid leukemias. In the 1 st 10 years of life siblings of patients with leukemia have a 4 -fold increased risk of leukemia with identical twins having a 20% chance of developing leukemia.

ETIOLOGY Etiology is unknown in most cases of leukemia. In humans there is strong evidence of a viral etiology for Adult T cell Leukemia/lymphoma associated with HTLV -I (geographically isolated to SW Japan, the Caribbean, and some sections of Africa). African Burkitt's lymphoma/leukemia is associated with the Epstein-Barr virus.

ETIOLOGY Environmental factors such as irradiation (e. g. increased incidence of leukemia in survivors of the atomic bomb explosions in Japan); early radiologists (9 fold increased risk); patients who have received therapeutic irradiation) chemical toxins (e. g. benzene, ), and drugs (e. g. chloramphenicol, phenylbutazone, and especially alkylating agent chemotherapy) have been associated with leukemia.

PATHOGENESIS From a morphologic and kinetic stand-point, there is a failure to differentiate and mature into functional hematopoietic cells. There is a loss of feed-back control, resulting in an over proliferation of immature and often functionally abnormal hematopoietic cells. Such cells may lack certain surface/cytoplasmic proteins and enzymes, i. e. . myeloperoxidase deficient, or produce aberrant products, i. e. . hemoglobin F. It is likely that leukemic cells produce cytokines or growth factors that inhibit normal hematopoietic growth.

PATHOGENESIS In most instances the leukemic cells remain functionally immature. Thus while retaining the capability to proliferate, they remain, for the most part, in the marrow space, replacing normal marrow elements.

SIGNS AND SYMPTOMS: Acute leukemia frequently presents with weakness, fatigue, fever, pallor, shortness of breath, weight loss, bone pain, night sweats and recurrent infections. The symptoms are often like the flu. Bruising, nosebleeds, unusally heavy menstrual periods, and swollen gums are commonly encountered in leukemia. Physical examination and laboratory studies may reveal anemia, splenomegaly, hepatomegaly and lymphadenopathy.

SIGNS AND SYMPTOMS: The chronic leukemias are often asymptomatic, but may present with relatively nonspecific and mild symptoms and signs. At later stages the signs and symptoms may be more like those of acute leukemia.

DIAGNOSIS AND PROGNOSIS It is important to distinguish acute leukemia from chronic leukemia as the prognosis and treatment approach differ greatly. Acute leukemias are generally treated aggressively with intensive chemotherapy while chronic leukemias are often watched or "followed" reserving chemotherapy for later stages when organ infiltration becomes a problem.

DIAGNOSIS AND PROGNOSIS It is important to distinguish myeloid from lymphoid for the type of chemotherapy used against acute lymphoblastic leukemia (vincristine, prednisone and anthracycline ± aspiraginase) is very different from the more highly toxic chemotherapy used for acute myeloid leukemia (cytosine arabinoside, anthracycline ± 6 thioguanine).

LABORATORY EVALUATION The diagnosis and classification of leukemia is accomplished largely through the laboratory examination of peripheral blood and bone marrow. The principle studies include: Morphology peripheral blood smear bone marrow aspirate & biopsy Cytochemistry Genetic analyses-chromosomal and molecular Immunology

AML Definition: AML involves the malignant proliferation of immature cells or blasts which are of nonlymphoid or myelogenous type. This proliferation originates in the bone marrow, but involves the peripheral blood and other organs. Diagnosis of AML: The presence of > 20% blasts in the BLOOD/ marrow as determined by a hematologist on PERIPHERAL SMEAR/bone marrow aspirate. Frequently see increased peripheral WBC count but it can be increased, normal or decreased.

WHO CLASSIFICATION OF AML l Acute myeloid leukaemias with recurrent cytogenetic abnormalities l Acute myeloid leukaemia with multilineage dysplasia l Acute myeloid leukaemia and myelo-dysplastic syndrome, therapy related l Acute myeloid leukaemia not otherwise categorised l Acute leukaemia of ambiguous lineage
(q 22;")
1. ACUTE MYELOID LEUKAEMIAS WITH RECURRENT CYTOGENETIC ABNORMALITIES l AML with t(8; 21)(q 22; q 22), (AML 1/ETO) l AML with inv(16)(p 13 q 22) or t(16; 16)(p 13; q 22), (CBFb/MYH 11) l Acute promyelocytic leukaemia [AM with t(15; 17)(q 22; q 12), (PML/ RARa) and variants] l AML with 11 q 23 (MLL) abnormalities

2. ACUTE MYELOID LEUKAEMIA WITH MULTILINEAGE DYSPLASIA l with prior myelo-dysplastic syndrome l without prior myelo-dysplastic syndrome

3. ACUTE MYELOID LEUKAEMIA AND MYELO-DYSPLASTIC SYNDROME, THERAPY RELATED l l Alkylating agent related Topoisomerase II inhibitor-related

4. ACUTE MYELOID LEUKAEMIA NOT OTHERWISE CATEGORISED l l l l l Acute myeloid leukaemia, minimally differentiated Acute myeloid leukaemia without maturation Acute myeloid leukaemia with maturation Acute myelomonocytic leukaemia Acute monoblastic and monocytic leukaemia Acute erythroid leukaemia Acute megakaryoblastic leukaemia Acute basophilic leukaemia Acute panmyelosis with myelofibrosis Myeloid sarcoma

MYELOBLASTS Myeloblasts have nuclei with fine, delicate chromatin and most often prominant nucleoli. The cytoplasm of myeloblasts tends to be moderate in volume and lightly basophilic without granules (primary azurophilic granules may be seen in myeloblasts).

AUER RODS IN MYELOBLASTS Type I myeloblasts have no azuriphilic primary granules nor Auer rods. Type II myeloblasts have a few ( <20) azuriphilic primary granules. Auer rods may be seen. Type III myeloblasts have >20 azuriphilic primary granules without a Golgi zone. Promyelocytes are larger, have a lower N/C ratio with denser chromatin, and usually have a pale paranuclear Golgi.

CYTOCHEMISTRY IN AML Myeloperoxidase M 1 M 2 MPO NSE PAS + - NSE M 3 + - / + - PAS M 4 M 5 + + - - / + ++ - / + M 6 M 7 + + + / +

LABORATORY DIAGNOSIS OF AML the definitive diagnosis of leukemia rests on the finding of increased blasts on bone marrow The white blood cell (WBC) count although usually high, is often normal or occassionally even low. The percent of blasts in the peripheral blood is highly variable and may range from 0 -100%. Immunophenotypic studies show expression of CD 13 and/or CD 33 on 95% of all nonlymphoid leukemias Chromosomal abnormalities are important diagnostic and prognostic findings. The most important are the t(8; 21) in M 2; t(15; 17) in M 3, and t(9; 11) in M 5.

MYELOID LEUKEMIA WITH MINIMAL DIFFERENTIATION - FAB M 0 dependent on the immunological identification of myeloid antigens. Morphologic and cytochemical studies show no distinguishing features.

ACUTE MYELOBLASTIC LEUKEMIA WITHOUT MATURATION - FAB M 1 > 90% type I and II blasts Most of the blasts are agranular. Auer rods are infrequent. Relatively few blasts (5 -10%) are MPO (myeloperoxidase) positive. A minimum of 3% MPO positive blasts are required for diagnosis. Variably positive for CD 13, CD 14, CD 11 b, CD 33, and HLA-DR. t(9; 22) Philadelphia chromosome, 8+, -5, and -7.
")
ACUTE MYELOBLASTIC LEUKEMIA WITH MATURATION - FAB M 2 is the most common (2040%) type of AML. Type II blasts are common and Auer rods are frequent The blasts are largely MPO positive. Variable positivity for CD 13, CD 33, and HLA-DR, but are negative for CD 14 and CD 11 b. t(8; 21), 8+, -5, and -7.
 - FAB M 3 the majority of the proliferating cells")
ACUTE PROMYELOCYTIC LEUKEMIA (APL) - FAB M 3 the majority of the proliferating cells are abnormal promyelocytes with numerous primary type granules. Auer rods are frequent and often multiple. The cells are MPO and chloroacetate esterase (CAE) positive, Positivity for CD 13 and CD 33, but are usually negative for HLA-DR. The t(15; 17) is unique to promyelocytic leukemia.

MICROGRANULAR VARIANT – M 3 the leukemic cells have a monocytic appearance with clefted angel-wing nuclei and abundant cytoplasm having at best indistinct cytoplasmic granulation. The cytochemical, immunophenotypic and chromosomal features are indentical to the classic M 3. Both forms of M 3 are associated with a high incidence of (DIC) and hemorrhage.
 - FAB M 4 myeloid and monocytic lines. Monocytes and")
ACUTE MYELOMONOCYTIC LEUKEMIA (AMML) - FAB M 4 myeloid and monocytic lines. Monocytes and promonocytes represent > 20%, but < 80% of the marrow differential. More than 20% of the blasts should be MPO + and more than 20% should be NSE +. High serum lysozyme (3 x normal) A peripheral monocytosis of t(4; 11), t(9; 11), 8+ and -7. M 4 e variant in which eosinophils (> 5%) are increased in number and abnormal associated with abnormalities of chromosome 16.
 - FAB M 5 Monocytic. Two subtypes: M 5 a")
ACUTE MONOCYTIC LEUKEMIA (AMOL) - FAB M 5 Monocytic. Two subtypes: M 5 a is the poorly differentiated form M 5 b is the welldifferentiated form t(9; 11), 8+, -5, and -7. Chromosome abnormalities of 11 q are closely associated with M 5 a

ACUTE ERYTHROBLASTIC LEUKEMIA - FAB M 6 or erythroleukemia is rare and difficult to diagnose. More than 30 - 50% of the nucleated marrow cells are abnormal nucleated red blood cells. The malignant red cells are PAS positive, Positive for glycophorin A. 8+, -5, del(5 q), and -7.
 - FAB M 7 blasts are often resemble lymphoblasts, may")
ACUTE MEGAKARYOCYTIC LEUKEMIA (AMKL) - FAB M 7 blasts are often resemble lymphoblasts, may be accompanied by atypical megakaryocytes. The marrow is often fibrotic. M 7 blasts are MPO negative and variably positive for PAS and NSE. ultracytochemical identification of platelet peroxidase (PPO). positive for glycoproteins GP Ib, GP IIb/IIIa, Factor VIII related protein - IHC

ADDITIONAL INFORMATION REGARDING LEUKEMIA Eosinophilic leukemia is a rare variant of acute myeloid leukemia in which blasts and immature eosinophils proliferate. CNS involvement appears to be common. Should be distinguished from CML with large numbers of eosinophils. Basophilic leukemia is a rare subset of AML associated with t(6; 9) and abnormal 12 p. Ultrastructural studies of the immature basophilic granules may be necessary. Philadelphia chromosome positive cases may be related to the blast crisis of CML.
 or granulocytic sarcoma or chloroma is an extramedullary tissue")
EXTRAMEDULLARY MYELOID CELL TUMOR (EMCT) or granulocytic sarcoma or chloroma is an extramedullary tissue mass of blasts and immature myeloid cells.

SUMMARY
- Slides: 37