LECTURE 12 SNPs LINKAGE POSITIONAL CLONING Mapping within




























































- Slides: 61
LECTURE #12 SNPs & LINKAGE: POSITIONAL CLONING
Mapping within a gene • can recombination happen within a gene? • in other words – do gene mutations change the whole gene at a single stroke? • or does a mutation change only a specific part of the gene where the mutation is located? • how do you locate these mutations? • these questions are answered by mapping within a gene
Seymour Benzer • used recombination analysis to show that two different mutations that don’t complement each other (i. e. make up for each other) are located on the same gene • proposed that XO can also occur within a gene = Intragenic Recombination • spent 10 years working on this project!!
Complementation Testing • cross-overs between homologous chromosomes with different mutations can generate: • 1. a wild-type allele AND • 2. a recombinant allele with both mutations • determined through Complementation Testing
T 4 bacteriophage • Benzer chose to work with the T 4 bacteriophage and the gene called r. II (r. II locus) • bacteriophage are viruses that infect bacteria • T 4 infects E. coli • head –haploid DNA genome of 200, 000 bp or 120 genes • 17 genes are essential to DNA replciation • 48 genes required for production of tail and tail fibers • remaining genes function in the life cycle in other ways
T 4 Life Cycle • infection – introduction of T 4 genome into bacterial cell • phage can only replicate in bacterial cells • bacteria stops making its own proteins and begins to make T 4 proteins • 30 minutes later – lysis and release of progeny T 4 (300 per bacterial cell) ~ 30 minutes 300 progeny per cell ~ 2 minutes ~ 6 minutes ~ 17 minutes
Advantages to using bacteriophage • each phage can produce 100 – 1000 progeny every hour (depending on environmental conditions) • easy to produce large numbers of progeny – allows for detection of rare genetic events • certain conditions can allow for the proliferation of only recombinant phages and the death of parental phages
T 4 Bacteriophage: Experimental Protocol • mix bacteria and bacteriophage and pour onto an agar plate – create a lawn of bacteria and phage • death of the bacteria because of the phage leaves a clear area on the plate called a plaque • the release of phage from one bacterial cell diffuses away to infect and kill neighboring bacteria producing the plaque • a typical plaque = 1 x 106 to 1 x 107 viral progeny
Benzer and the T 4 Complementation assay • Benzer created T 4 mutants that produced large plaques because of abnormally rapid lysis of bacterial host cells • called these mutations ‘r’ for rapid • many of these r mutations mapped to the r. II locus in the phage genome • phage with mutations – r. II • wild type phage – r. II+ • infected two strains of E. coli – B and K(l) • observed different phenotypes depending on the strain infected note that r. II- phage cannot form plaques in K(l) bacteria
Benzer and the T 4 Complementation assay • but before Benzer could perform his experiments on recombination – he had to confirm that when he infected E. coli – two T 4 phages entered the host cell • used a simple complementation test using two different types of T 4 phage • used these phage to identify two complementation groups (i. e. genes) in the r. II locus: • group r. IIA – mutations in gene r. IIA • group r. II B – mutations in gene r. IIB r. IIA • used the K(l) bacteria because only a wild type r. II gene can cause lysis in this bacterial strain r. IIAA r. IIB B
Benzer and the T 4 Complementation assay • reasoning: if the two mutations were on different genes – recombination would regenerate a wild-type allele – bacterial lysis of K(l) would result = plaques r. IIA r. IIB
Benzer and the T 4 Complementation assay • if they were on the same gene – no lysis and plaques would result r. IIB
Can Recombination occur between two mutations in a single gene? • having confirmed that he could use two T 4 phages and get them both to infect a single bacterial cell • used two T 4 phages BUT these phages had different mutations IN THE SAME GENE • called these r. II- mutations r. IIA 1 and r. IIA 2 • devised a simple and elegant test based on his observation that two T 4 genomes must enter a bacterial cell for complementation to happen between these two mutations • used the two strains of E. col called B and K(l) • r. II- mutant bacteriophages cannot form plaques in the K(l) strain
Can Recombination occur between two mutations in a single gene? • REASONING: if recombination takes place within a gene – then it will generate a wild type allele (with no r. II mutations) and a mutated allele with both r. IIA 1 and r. IIA 2 mutations • those bacteriophage with the wild type allele can lyse K(l) bacteria and produce plaques • those bacteriophage with both r. IIA mutations cannot lyse the bacteria and produce plaques • the presence of plaques allowed Benzer to confirm that recombination can happen within a gene
Can Recombination occur between two mutations in a single gene? • as a control: he infected K(l) bacteria with only one of the two T 4 mutant strains • proving that T 4 with mutated r. IIA genes are unable to lyse K(l) bacteria and produce plaques
Trans vs. Cis • complementation test on T 4 showed two things: • 2 mutants were in two different genes = trans configuration • 2 mutants on the same gene = cis configuration • complementation tests are also known as a cis-trans test • Benzer called any complementation group identified by this test = cistron • often used synonomously for gene
Using deletions to map mutations on the same gene • cross a bacteriophage with a mutation with a bacteriophage with a deletion in that region • scan slide • if point mutation is located in the same region as the deletion – no recombination is possible no plaques • if the point mutation lies outside the deletion – recombination can occur
Using deletions to map mutations on the same gene • crosses between an uncharacterized mutation and a known deletion will reveal where this mutation is = Deletion Mapping
Benzer’s Strategy for Fine Mapping • fine mapping – genes are made up of discrete units arranged in a linear fashion in a small portion of a chromosome • his strategy: • 1. co-infect mutants to define complementation groups (genes) • determine relationships between deletions • 2. co-infect each r. II- mutation with deletions to group them • to localize the point mutation • 3. look for recombination between mutations in the same deletion region • produces a “fine map” of a specific gene
Deletion Mapping of the r. II locus • Benzer divided the r. II regions into a series of intervals • assigned a mutation to an interval by looking to see if recombination occurred to give the wild type allele and infection/plaque formation • mapped 1612 mutations and several deletions • hot spots = regions that spontaneously mutate more frequently
Take a Break NEXT TOPIC: LINKAGE ANALYSIS IN HUMANS – POSITIONAL CLONING
Detecting a mutation within a gene • cystic fibrosis • in vitro fertilization • blastocyst – remove a cell • PCR to amplify chromosome 7 • perform a Southern Blot • run the PCR product on an agarose gel and transfer to a nitrocellulose membrane • expose the membrane to 2 probes: • 1. wild type sequence • 2. mutant sequence • alternatively – could PCR the region known to have the CFTR mutation • but what if you don’t know where the mutation is exactly on a gene? ?
Genotyping • genotyping = to detect which alleles are present in the donor cells • geneticists use an array of molecular tools to detect DNA differences among individuals • e. g. PCR amplification of regions of genome • DNA genotyping can then predict the possibility of a disease
Strategies for Analyzing Genomes • 1. Genotyping protocols • DNA fingerprinting, PCR, microarrays • 2. Positional cloning • application of genotyping protocols to linkage analysis in an organism • 1. mapping • 2. cloning • 3. mutation screening • 3. Haplotype associated studies • mapping of a disease locus in humans • relies upon the chromosome’s evolutionary history
Positional Cloning Key Concepts • locating a gene to a location on a chromosome – Positional Cloning • PC is the method of choice for identifying genetic mutations underlying diseases with simple Mendelian inheritance • it is a method of gene identification in which a gene is identifying only by its approximate chromosomal location • the candidate region for the gene is initially ID’d by linkage analysis • PC is then used to narrow the region
Positional Cloning Key Concepts • how does linkage mapping work in positional cloning? • remember linked alleles co-segregate • co-segregation increases as map distance decreases • the frequency of recombination is proportional to map distance – RF increases as map distance increases • strategy: pinpoint the gene’s location on a chromosome by determining which nearby alleles segregate with it • the more often they segregate together – the closer the gene is to that allele’s region on the chromosome • PC is very effective for locating disease genes with Mendelian inheritance • e. g. muscular dystrophy, cystic fibrosis, Huntington’s disease
Positional Cloning Key Concepts • PROBLEM: finding non-disease alleles in humans that are single trait genes with straightforward Mendelian inheritance • not enough phenotypically evident single gene traits with which to analyze linkage • most phenotypes in humans are caused by more than one gene trait • PROBLEM: can’t do two and three-point test crosses in humans • BUT the entire human genome has been sequenced • can now map with respect to genetic variations that don’t cause a visible phenotype • e. g. map with respect to a single nucleotide polymorphism or SNP
Positional Cloning in Humans • in humans – link a gene to genetic variations known as polymorphisms
Polymorphisms • members of the same species show enormous DNA variation in their genomes • 250 kb region of the CFTR gene • 1 difference every 1000 bp • 250 differences total • of the 250 kb – only 4. 0 kb of this region (2%) is the exons that code for the CFTR protein • mutations in these exons are negative mutations and are not usually passed on • variations in the intron sequences are passed on – little to no effect on the health of the individual
Polymorphisms • variations at a position in the genome can be considered as an alternate allele of a specific locus • originally studied using breeding and mutation studies • now studied molecularly • two or more alleles at a specific locus = polymorphic locus • variations = DNA polymorphisms • for genotyping – are 5 classes of DNA polymorphisms: • 1. SNP – single nucleotide polymorphisms • 2. microsatellite & minisatellite DNA – simple sequence repeats (SSRs) or small tandem repeats (STRs) • 3. Insertion/Deletions or DIPs • 4. Copy # variation (CNV)/Copy # polymorphisms - VNTRs • 5. Complex Variants
Polymorphism SNP DIP SSR/microsatellite Copy # variant Size Frequency – 1 per…. 1 bp 1 kb 1 to 100 bp 10 kb 1 to 10 bp repeats 30 kb 10 bp to 1 Mb 3 Mb
SNPs • single nucleotide polymorphisms • most prevalent type of polymorphism • about 1/700 base pairs differ in the human genome • arise from a mutation of a single base pair • errors in DNA replication or from a mutagen • still a very low mutation rate – 1 in 30 million bases • can be located anywhere in the genome – inside and outside of gene • bi-allelic – have two forms (maternal and paternal) • over 50 million SNPs known to date • 15 million of them are human • database - http: //www. ncbi. nlm. nih. gov/SNP/ • most have no phenotypic effect
SNPs
SNP Variations • most SNP variations in humans are confined to a limited number of positions • e. g. genomes of Craig Venter and James Watson and an anonymous donor were analyzed for known human SNPs • each of the three men have over one million unique SNPs • 2. 6 million SNPs were shared either by two or all three of them • most were “silent” SNPs – no effect on genes • 5000 SNPs had an effect on the amino acid sequence of a protein
SNP Variations • chromosome 7 – 400 kb region (base pair 116, 700, 001 to 117, 100, 000) • vertical lines represent the locations of the SNPs • all SNPs = human data base of SNPs • 3. 3 million used SNPs to distinguish the two genomes • 82% of known human SNPs (i. e. all SNPs) found in Venter • 86% of known human SNPs found in Watson • 20 kb block of SNPs found to be in common between the two men and the human database
Detecting SNPs • How are SNPs be detected? • 1. Sequencing – expensive • 2. Restriction Fragment Length Polymorphisms (RFLP) • relies on the use of restriction enzymes that cut DNA sequences at specific sequences • Restriction Enzyme – isolated from bacteria • used by bacteria to “cut-up” invading DNA – e. g. from bacteriophages • recognizes a unique sequence • the RE cuts the phosphodiester bond between two nucleotides within that sequence • produces either “sticky” ends or “blunt” ends
RFLP Analysis • several Eco. RI sites located within the human genome • cutting the genome gives a specific pattern of DNA fragments when run on an agarose gel • if one Eco. RI site has an SNP – the RE will no longer cut • OR an SNP could create an extra Eco. RI site in the genome • run the results of the restriction enzyme digest on an agarose gel • polymorphisms result in a unique banding pattern for each DNA sample
RFLP Analysis • three vs. two restriction enzyme sites in a genome • allele 1 – three sites 4 fragments • allele 2 has lost one site due to an SNP 3 fragments
RFLP Analysis & the Southern Blot • fragments are separated by size on an agarose gel • the DNA is transferred to a positively charged membrane overnight (through simple attraction of –ve DNA to the +ve membrane) • the membrane is then incubated with “probes” to well-known SNP sequences • the probes hybridize to the bands on the membrane and are easily visualized
Jack and Jill and RFLP • Jack and Jill – section of the genome with two Eco. RI sites • cut their DNA samples with Eco. RI • since Jack and Jill are diploid – they have two forms of this section of DNA – i. e. two alleles • the allele shown below – they are identical Jack 1: -GAATTC---(8. 2 kb)---GCATGCATGCAT---(4. 2 kb)---GAATTCJill 1: -GAATTC---(8. 2 kb)---GCATGCATGCAT---(4. 2 kb)---GAATTC-
Jack and Jill and RFLP • but this allele – they are NOT • Jack is missing the Eco. RI site on the left side of the genomic fragment Jack 2: -CCCTTC---(8. 2 kb)---GCATGCATGCAT---(4. 2 kb)---GAATTCJill 2: -GAATTC---(8. 2 kb)---GCATGCATGCAT---(4. 2 kb)---GAATTC- • therefore, when Jack and Jill have their DNA subject to RFLP analysis, they will have one band in common and one band that does not match the other's in molecular weight:
Jack and Jill and RFLP • to easily identify the differences – Southern blotting using a probe to a piece of DNA that lies between the two Eco. RI sites • because Jack is missing his “left-side” Eco. RI – he will have a larger DNA fragment than Jill and the probe will make it easy to see on the gel
Detecting SNPs • more modern methods of detecting SNPs now exist • allow for the detection of millions of SNPs • 3. Microarrays: • detection of SNP alleles at over 1 million loci in the human genome • freely accessible database at the NCBI
SNPs and Positional Cloning of a Disease-causing gene • GOAL: locate the position of a disease-causing gene in the human genome • WHY? • better basic understanding of the disease process • better diagnosis of the disease • design of more specific treatments • gene therapy to “cure” the disease
SNPs and Positional Cloning of a Disease-causing gene • STRATEGY: collect the DNA from populations in which the disease has been characterized • easiest diseases to positionally clone are Mendelian • create a pedigree chart • look for alleles that are found in diseases individuals more often than by chance – Linkage analysis • a lot like locating the “eye size” gene in Drosophila • indicates that the disease gene lies nearby • narrows the location to a chromosome or even a region on a chromosome • use linkage analysis & SNPs to narrow the region even further
SNPs and Positional Cloning of a Disease-causing gene • because SNPs are scattered throughout the human genome - they make it possible to test linkage to a gene with virtually any genomic location • APPROACH: • to effectively locate the disease gene – should use SNPs that are located every 10 centi. Morgans (or map • • • units) in the human genome – a c. M is about 1, 000 bp the human genome is 3000 c. M – so 300 SNPs will “cover” the entire genome of these 300 – find the SNP alleles that segregate with the disease more often than by chance these are the linked SNPs if they are not linked then the SNP and disease gene will segregate with equal frequency in diseased and non-diseased individuals
SNPs and Positional Cloning of a Disease-causing gene • once the disease gene has been localized to a smaller region of a chromosome – use SNPs in that smaller region • genotype the individuals of the pedigree (diseased and non-diseased) for the SNPs in this region • determine if these new SNPs are linked SNPs • if there is less than 1% recombination between the SNP and the disease locus – then the gene is less than 1 c. M away
SNPs and Positional Cloning of a Disease-causing gene • continue to narrow down the region until you find the SNPs that show 100% linkage with the disease • you have identified its specific location on the chromosome • this entire process of narrowing down is known as Positional Cloning
Huntington’s Disease • 1 st gene to be positionally cloned • 1984
Mapping – the X chromosome
Other DNA polymorphisms: DIPs • deletion-insertion polymorphisms • also know as In. Dels • typically a few base pairs in length • 2 nd most common form of genetic variation • result from errors in replication or DNA repair • if they occur in a protein coding region – can produce a shift in reading frame • e. g. Venter genome vs. human reference seq database – 292, 102 unique DIPs • 1 bp to 571 bps • as the length of the DIP increases, their frequency decreases • e. g. CFTR gene – DIPs once every 10 kb of DNA • -75% are only 1 to 2 base pairs
Other DNA polymorphisms: SSRs • simple sequence repeats or microsatellite DNA • also known as small tandem repeat (STRs) • arise by random events that produce a short repetitive sequence – 4 to 5 bp units repeated • can be highly polymorphic in the number of repeats – repeats from 10 to 100 times • produces alleles • e. g. maternal allele – 10 repeats • paternal allele – 25 repeats • produced by the “stuttering” of the DNA polymerase during replication • one of the most common SSR is a 2 bp repeat – “CA” repeat
Other DNA polymorphisms: SSRs • once they form – they can lengthen by the “stuttering” of the DNA polymerase during replication • DNA polymerase pauses - top daughter strand slips and produces a loop • replication continues and the daughter strand is replicated • during the next round of replication when this daughter strand is the template – the “straightening” out makes for a longer template and the resulting template/daughter ds helix now has an increased # of repeats
SSRs can be detected by PCR • SSR alleles differs in length • e. g. maternal allele 1 – 15 repeats • paternal allele 2 – 35 repeats • PCR using primers that flank these SSR regions • size differences easy to see on a gel • larger SSR allele won’t run as far into the agarose gel
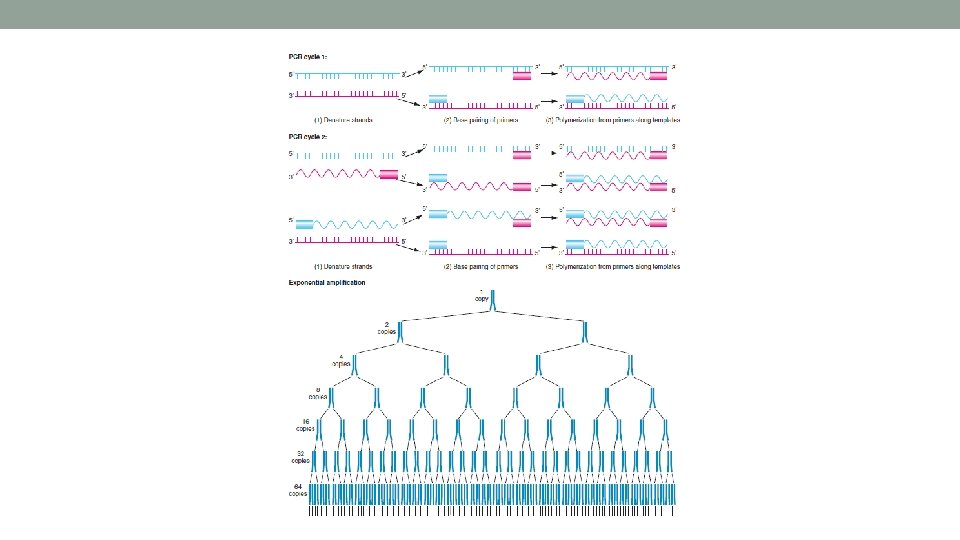
Polymerase Chain Reaction PCR • used to amplify specific regions of DNA • mimics DNA replication that takes place in cells • uses temperature to mimic some of the enzymatic steps • e. g. heat ds DNA to 94 C to “melt” or denature the two strands – mimics the helicase • uses custom designed primers that target specific DNA sequences • takes the place of primase making them • polymerase = Taq polymerase • heat resistant polymerase from bacteria (Thermus aquaticus)
PCR Primers • primers are designed to hybridize with (i. e. to • • • “anneal”) to the template DNA primers “flank” the target region you want to amplify forward primer (primer 1) - anneals to the antisense template reverse primer (primer 2) – anneals to the sensetemplate strand annealing takes place when the PCR reaction is cooled from 94 C to a specific annealing temperature – usually between 55 and 65 C the polymerase binds to the template-primer double stranded region – just like it would inside the nucleus of a cell
Taq polymerase • heat resistant bacterial polymerase • capable of withstanding high denaturing temperatures like 94 C • binds to the primer-template doubled stranded complex and “extends” from it • moves along the template strand in the 3’ to 5’ direction – making “daughter” DNA that grows in the 5’ to 3’ direction • this extension step occurs at 72 C
The PCR Reaction • PCR is performed over multiple “cycles” CYCLE 1 • one cycle has three temperatures • 1. Denaturing = 94 C • 2. Annealing = 55 to 65 C • 3. Extension = 72 C original template daughter DNA original template • after the 1 st cycle – you have two ds DNA strands original template • 2 nd cycle – DNA denatures 4 single DNA strands that the primers anneal to • amplification of the targeted region begins • after 2 cycles – 4 ds DNA strands daughter template original template • multiple cycles result in the amplification of the targeted DNA CYCLE 2 original template daughter DNA daughter template original template
SSRs can be detected by PCR • SSR analysis produces a DNA “fingerprint”