Last lecture summary identity vs similarity homology vs



 Pam 40 Short highly similar alignments")
 • Recursive approach, sequential dependency. • 4 th piece can be")






 • Stringency")

 –")

















- Slides: 33
Last lecture summary
• identity vs. similarity • homology vs. similarity • gap penalty • affine gap penalty • gap penalty high • fewer gaps, if investigating related sequences • low • more gaps, larger gaps, distantly related sequences
BLOSUM • blocks • focuse on substitution patterns only in blocks • BLOSUM 62 – 62, what does it mean? • BLOSUM vs. PAM • BLOSUM matrices are based on observed alignments • BLOSUM numbering system goes in reversing order as the PAM numbering system
Selecting an Appropriate Matrix Best use Similarity (%) Pam 40 Short highly similar alignments 70 -90 PAM 160 Detecting members of a protein family 50 -60 PAM 250 Longer alingments of more divergent sequences ~30 BLOSUM 90 Short highly similar alignments 70 -90 BLOSUM 80 Detecting members of a protein family 50 -60 BLOSUM 62 Most effective in finding all potential similarities 30 -40 BLOSUM 30 Longer alingments of more divergent sequences <30 Similarity column gives range of similarities that the matrix is able to best detect.
Dynamic programming (DP) • Recursive approach, sequential dependency. • 4 th piece can be solved using solution of the 3 rd piece, the 3 rd piece can be solved by using solution of the 2 nd piece and so on…
New best alignment = previous best + local best Best previous alignment Sequence A. . . Sequence B If you already have the optimal solution to: X…Y A…B then you know the next pair of characters will either be: X…YZ A…BC or X…YZ A…B- You can extend the match by determining which of these has the highest score.
New stuff
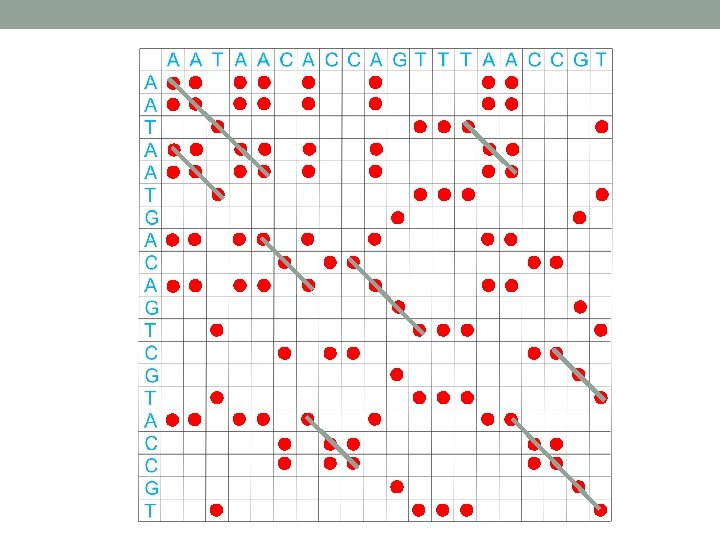
Dot plot • Graphical method that allows the comparison of two biological sequences and identify regions of close similarity between them. • Also used for finding direct or inverted repeats in sequences. • Or for prediction regions in RNA that are selfcomplementary and therefore have potential to form secondary structures.
Self-similarity dot plot I The DNA sequence EU 127468. 1 compared against itself. Introduction to dot-plots, Jan Schulz http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
background noise gap runs of matched residues
Self-similarity dot plot II The DNA sequence EU 127468. 1 compared against itself. Window size = 16. Linear color mapping Introduction to dot-plots, Jan Schulz http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
Improving dot plot • Sliding window – window size (lets say 11) • Stringency (lets say 7) – a dot is printed only if 7 out of the next 11 positions in the sequence are identical • Color mapping • Scoring matrices can be used to assign a score to each substitution. These numbers then can be converted to gray/color.
Interpretation of dot plot I 1. Plot two homologous sequences of interest. If they similar – diagonal line will occur (matches). 2. frame shifts a) mutations gaps in diagonal b) insertions shift of main diagonal c) deletions shift of main diagonal http: //ugene. unipro. ru/documentation/manual/plugins/dotplot/interpret_a_dotplot. html
Interpretation of dot plot II • Identify repeat regions (direct repeats, inverted repeats) – lines parallel to the diagonal line in self-similarity plot • Microsattelites and minisattelites (these are also called low-complexity regions) can be identified as “squares”. • Palindromatic sequences are shown as lines perpendicular to the main diagonal. • Plaindromatic sequence: V ELIPSE SPI LEV Bioinformatics explained: Dot plots, http: //www. clcbio. com/index. php? id=1330&manual=BE_Dot_plots. html
Repeats in dot plot minisattelites self-similarity dot plot of NA sequence ofhuman LDL receptor window 23, stringency 7 direct repeats inverted repeats from the book Bioinformatics, David. M. Mount,
Interpretation of dot plot – summary http: //www. code 10. info/index. php? option=com_content&view=article&id=64: inroduction-to-dot-plots&catid=52: cat_coding_algorithms_dot-plots&Itemid=76
Dot plot of the human genome A. M. Campbell, L. J. Heyer, Discovering genomics, proteomics and bioinformatics
Dot plot rules • Larger windows size is used for DNA sequences because the number of random matches is much greater due to the presence of only four characters in the alphabet. • A typical window size for DNA is 15, with stringency 10. For proteins the matrix has not to be filtered at all, or windows 2/3 with stringency 2 can be used. • If two proteins are expected to be related but to have long regions of dissimilar sequence with only a small proportion of identities, such as similar active sites, a large window, e. g. , 20, and small stringency, e. g. , 5, should be useful for seeing any similarity.
Dot plot advantages/disadvantages • Advantages: • All possible matches of residues between two sequences are found. It’s just up to you to choose the most significant ones. • Readily reveals the presence of insertions/deletions and direct and inverted repeats that are more difficult to find by the other, more automated methods. • Disadvantages: Most dot matrix computer programs do not show an actual alignment. Does not return a score to indicate how ‘optimal’ a given alignment is (no statistical significance that could be tested).
Homology vs. similarity again • Just a reminder of the important concept in sequence analysis – homology. It is a conclusion about a common ancestral relationship drawn from sequence similarity. • Sequence similarity is a direct result of observation from the sequence alignment. It can be quantified using percentages, but homology can not! • It is important to understand this difference between homology and similarity. • If the similarity is high enough, a common evolutionary relationship can be inferred.
Limits of detection of alignment • However, what is enough? What are the detection limits of pairwise alignments? How many mutations can occur before the differences make two sequences unrecognizable? • Intuitively, at some point are two homologous sequences too divergent for their alignment to be recognized as significant. • The best way to determine detection limits of pairwise alignment is to use statistical hypothesis testing. See later.
Twilight zone • However, the level one can infer homologous relationship depends on type of sequence (proteins, NA) and on the length of the alignment. • Unrelated sequences of DNA have at least 25% chance to be identical. For proteins it is 5%. If gaps are allowed, this percentage can increase up to 10 -20%. • The shorter the sequence, the higher the chance that some alignment is attributable to random chance. • This suggest that shorter sequences require higher cuttof for inferring homology than longer sequences.
Essential bioinformatics, Xiong
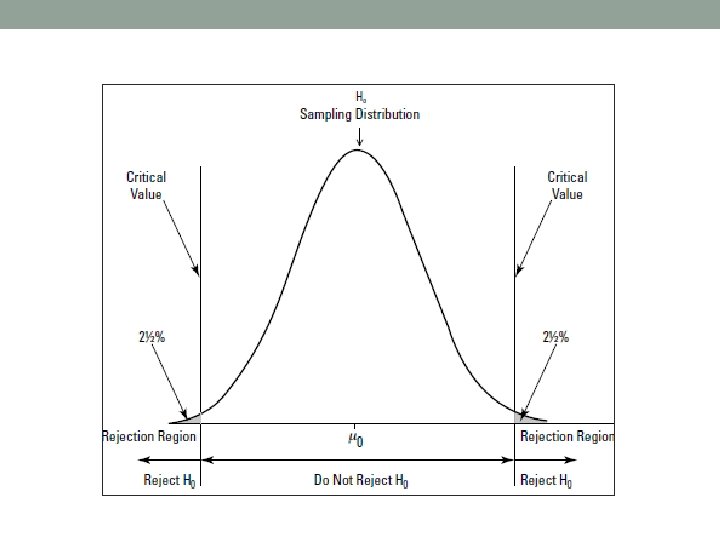
Statistical significance • Key question – Constitutes a given alignment evidence for homology? Or did it occur just by chance? • The statistical significance of the alignment (i. e. its score) can be tested by statistical hypotheses testing. • The matched sequence reported e. g. by the search program can be classified as TP (true positive, i. e. two sequences are homologous) or as FP (false positive, i. e. genuinely unrelated, aligned only by chance).
Significance of global alignment I •
Significance of global alignment II •
Significance of global alignment III •
Significance of global alignment IV •
Significance of local alignment • In contrast to global alignment there is a thorough understanding of the distribution of scores. • Key role play Extreme value distributions (EVD) • generate N data sets from the same distribution, create a new data set that includes the maximum/minimum values from these N data sets, the resulting data set can only be described by one of the three distributions • Gumbel, Fréchet, Weibull • applications • extreme floods, large wildfires • large insurance losses • size of freak waves • sequence alignment
Gumbel distribution wikipedia. org
Statistical distribution of alignments • local alignment • analytical theory • gapless – Gumbel, parameters can be evaluated analytically • gapped – Gumbel, paramaters must be obtained from simulations, no analytical formulas • global alignment • no thorough theory, however empirical simultions show that the distribution is also Gumbel