Kocaeli niversitesi Tp Fakltesi ocuk Sal ve Hastalklar






 • Boy: 102")


 • • Akraba evliliği öyküsü")

 • Lizozomal dışı nörometabolik hastalıklar")

 Saudubray JM, van den Berghe G, Walter JH. Inborn")

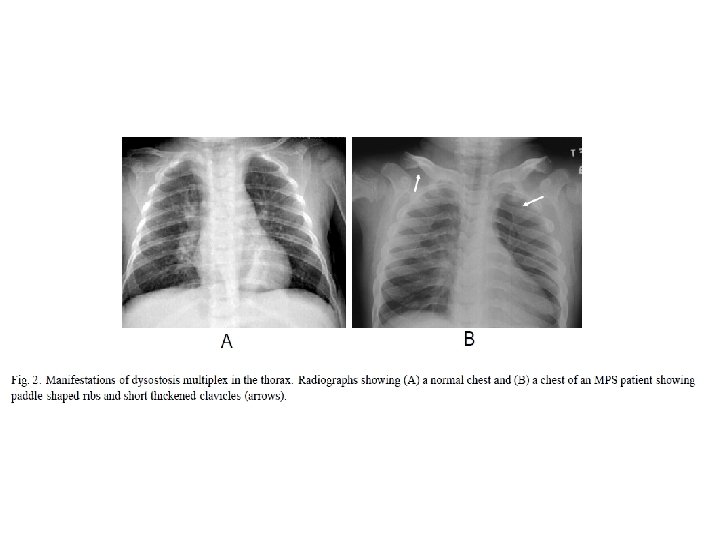
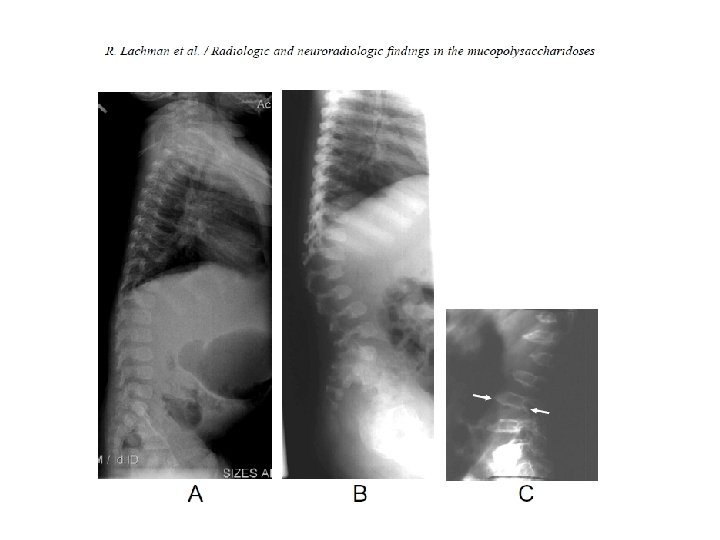
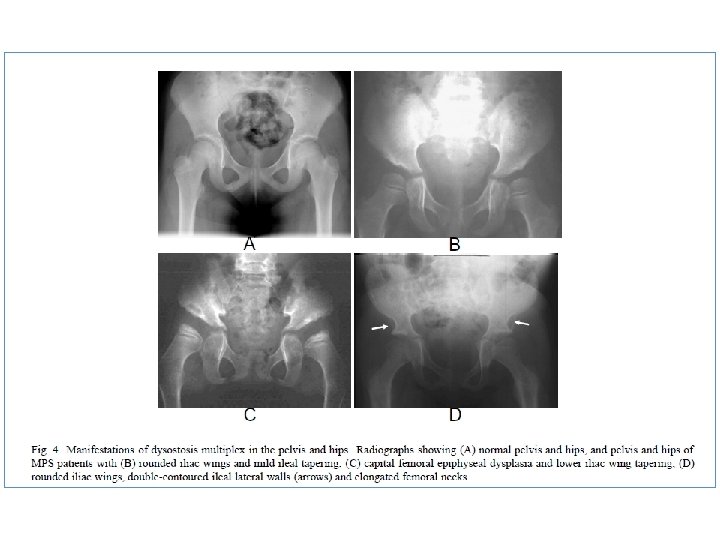

 • • Oligosakkarid: Trisakkarid bandı izlendi.")


")
: Genellikle 10 yaşından sonra")
 q. Kemik")
 q.")




- Slides: 40
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları Anabilim Dalı Nöroloji Bilim Dalı Olgu Sunumu 19 Aralık 2014 Cuma Uzm. Dr. Emek Uyur Yalçın Uzm. Dr. Hülya Maraş Genç Prof. Dr. Bülent Kara
19/12/2014 ÇOCUK NÖROLOJİ BİLİM DALI OLGU SUNUMU Dr. Emek Uyur Yalçın Dr. Hülya Maraş Genç Dr. Bülent Kara
Olgu • 4 yaş 4/12 aylık erkek hasta • Şikayeti: Yürümede gecikme, konuşamama. • Öyküsü: Konuşmada gecikme nedeni ile başvurduğu merkezde, KBB muayenesi ve işitme testi yapılması önerilmiş. İzlemde tekrarlayan seröz otit nedeni ile her iki kulağa timpanostomi tüpü takılmış. Hasta yürümesinde de gecikmesi olması üzerine tarafımıza yönlendirildi.
Özgeçmiş • Prenatal : Doktor kontrolünde düzenli USG takibi yapılmış, sigara-alkol-ilaç kullanımı öyküsü yok, enfeksiyon-gestasyonel DMpreeklampsi öyküsü yok, X-ray maruziyeti yok. • Natal: Term AGA ve 3200 gram olarak NSVD ile doğmuş. • Postnatal : Doğar doğmaz ağlamış, siyanozu olmamış, hipoglisemi yaşamamış, YD sarılığı geçirmemiş. • Hidrosel nedeni ile 1 yaşında opere edilmiş.
Soygeçmiş • Anne: 26 yaşında, sağ-sağlıklı, ev hanımı • Baba: 28 yaşında, sağ-sağlıklı, işçi. • Akraba evliliği var. (2. 5 derece kuzen) • İlk gebelik düşükle sonuçlanmış, 2. gebelikten olgumuz doğmuş. • Anne halen 3 haftalık gebe.
Nöromotor gelişimi Ø Baş tutma: 4 ay Ø Destekli oturma: 9 ay Ø Desteksiz oturma: 12 ay Ø Heceleme: 3 yaş / Şu an 3 -4 kelimesi var, cümle kuramıyor. Ø Yürüme : 2. 5 yaş
Fizik Muayene • Baş çevresi: 52. 5 cm (75 -90. p) • Boy: 102 cm (50 -75. p) • Tartı: 21 kg (90 -97 p. )
Fizik Muayene • Bilinci açık, kısmen koopere, göz kontağı kuruyor, komutları alıyor, ortak dikkati var. • Tonusu normal, DTR’leri hiperaktif, klonusu yok, plantar yanıtı bilateral fleksör. Kas gücü muayenesine koopere olamadı. • Kaba yüz görünümü, dişeti hipertrofisi, belirgin alın, burun kökü basıklığı, hafif prognatizm mevcut. • Dalak kot altında ele geliyor, traube kapalı. • Diğer sistem muayeneleri doğal. • Şaşılık nedeni ile gözlük kullanıyor.
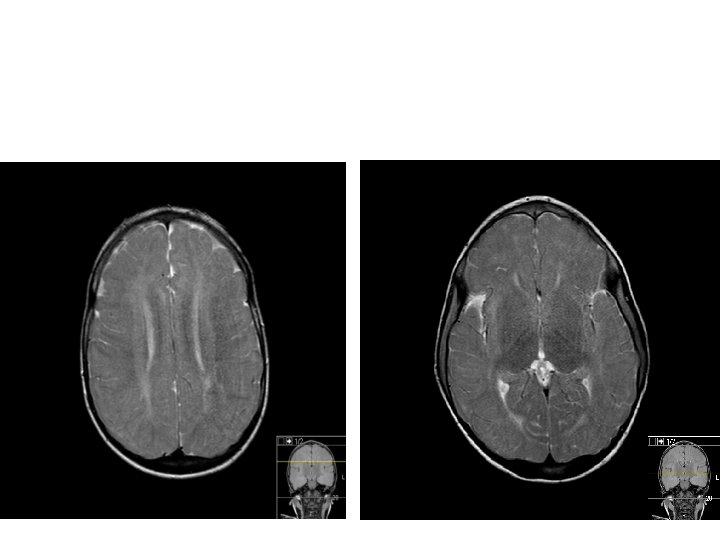
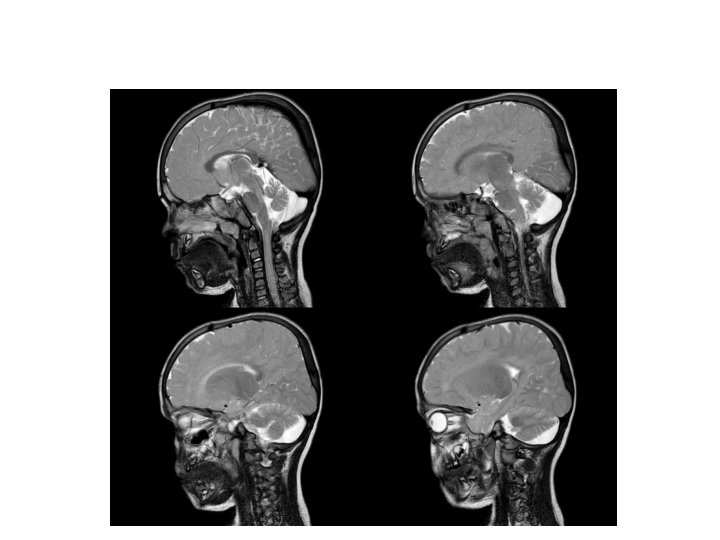
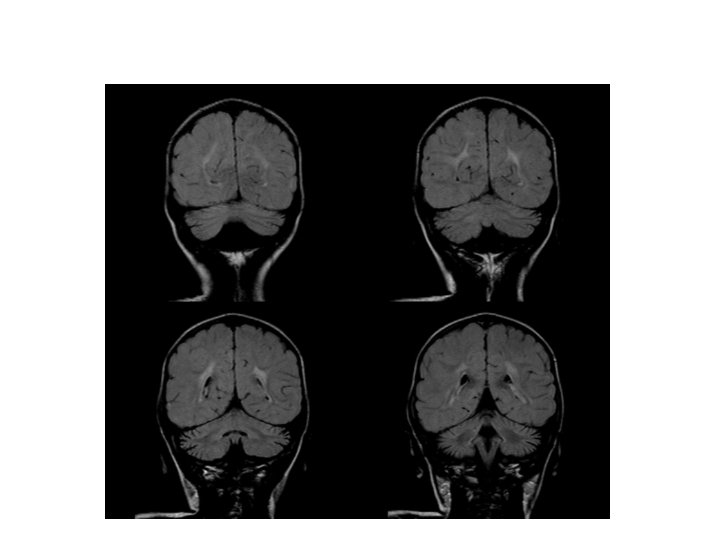
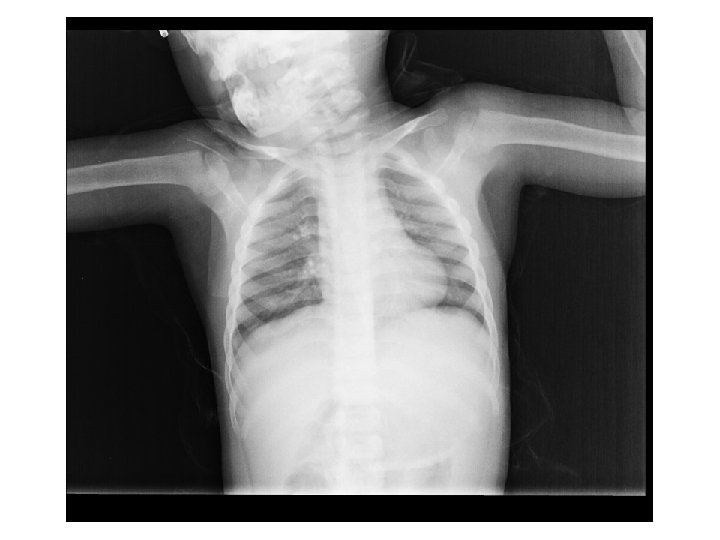
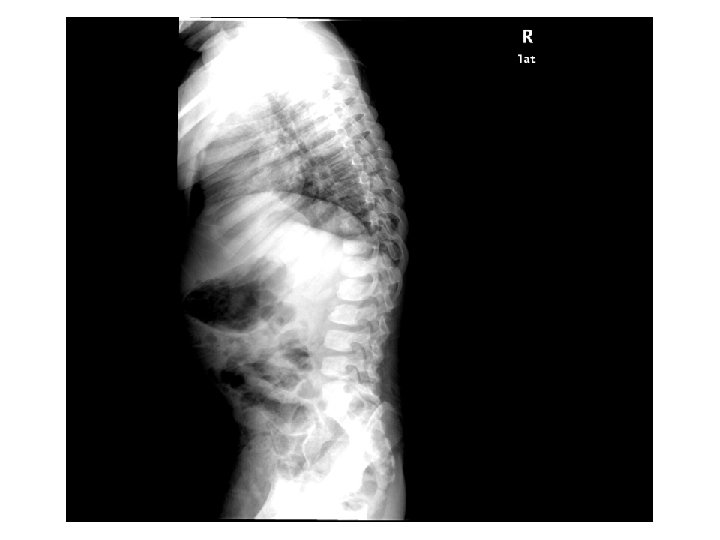
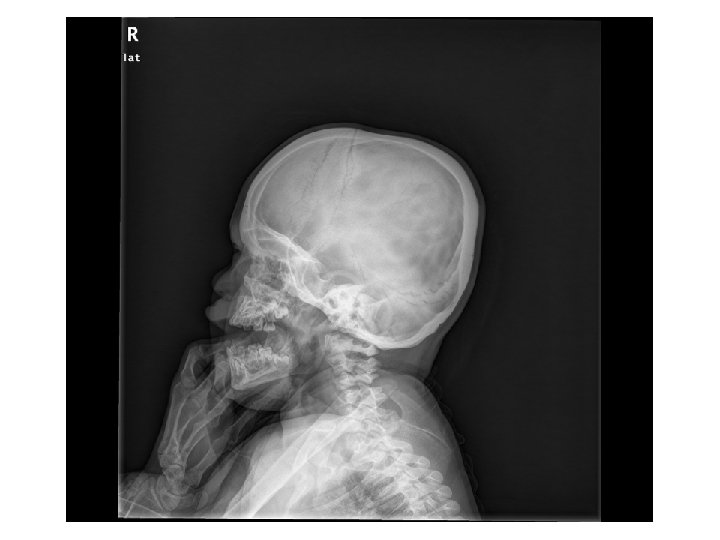
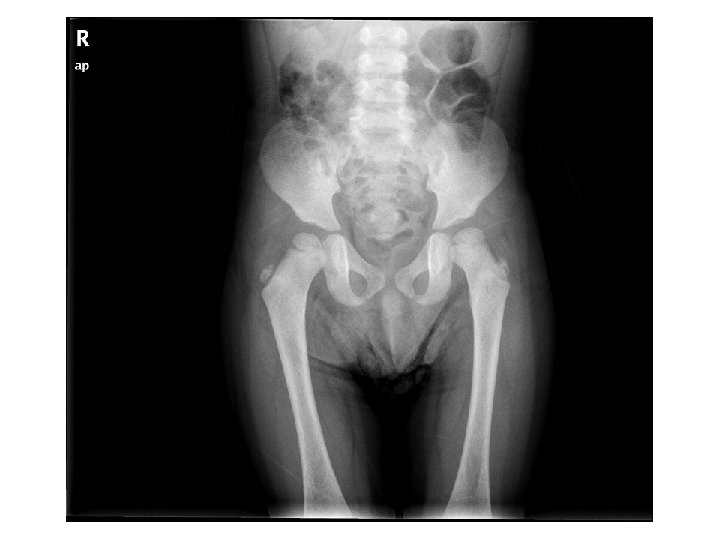
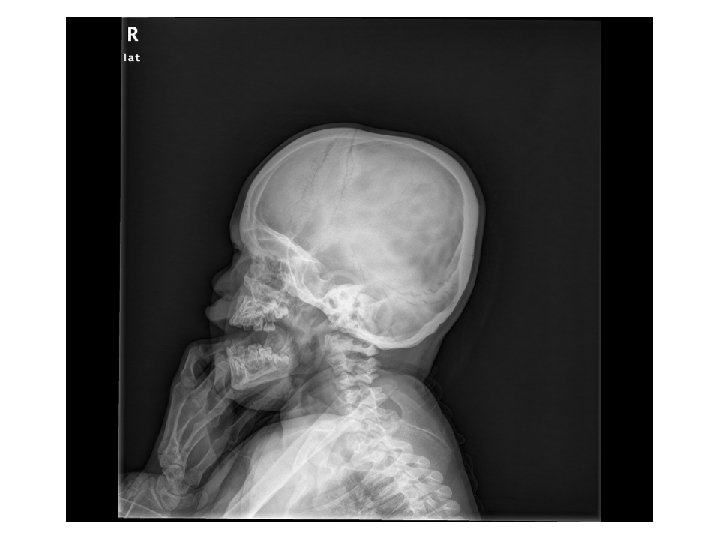
Tetkikler Hemogram, genişletilmiş biyokimya, CPK Amonyak, laktat, kan gazı Normal Tiroid fonksiyon testleri, B 12, folat TANDEM MS/MS İdrarda organik asit analizi EKO: Aort stenozu (valvuler-hafif) İşitme testi: Bilateral işitme kaybı Göz muayenesi: Strabismus Kranyal MRG’sinde (mayıs 2014); bilateral korona radiata ve tragonda, sağ temporalde T 2 hiperintensiteler ve serebellar atrofi. • İskelet grafileri • • •
Olağan dışı bulgular (4 yaş 4 aylık erkek hasta) • • Akraba evliliği öyküsü Tekrarlayan seröz otit atakları ve işitme kaybı Global gelişme geriliği (konuşma geriliği belirgin) Kaba yüz görünümü, dişeti hipertrofisi, belirgin alın, burun kökü basıklığı, hafif prognatizm Splenomegali Aort stenozu (valvuler-hafif) ‘’Dizostozis multipleks’’ Şaşılık
• ÖN TANI?
Ayırıcı Tanı • Lizozomal depo hastalıkları (özellikle MPS grubu) • Lizozomal dışı nörometabolik hastalıklar
‘‘Dizostozis Multipleks’’in görüldüğü depo hastalıkları • Mukopolisakkaridozlar • Mukolipidozlar (I-Cell hastalığı ve Pseudo. Hurler Polidistrofisi) • Oligosakkaridozlar • Multipl sulfataz eksikliği • Konjenital glikozilasyon defektleri • GM I gangliozidoz
Hurler sendromu (MPS tip 1) Saudubray JM, van den Berghe G, Walter JH. Inborn Metabolic Diseases Diagnosis and Treatment. 5 th edition. Heidelberg. 2012: 581
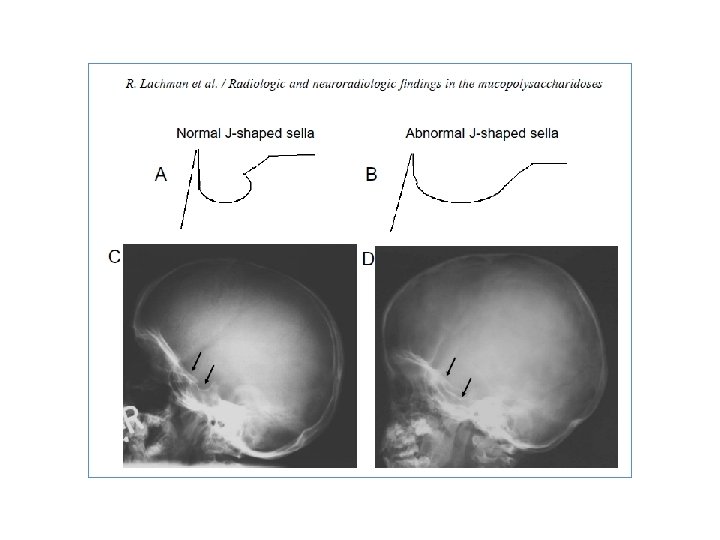
Metakarpal kemiklerin proksimal bölümlerinde düzensizlik ve noktalanma Courtesy of Ed Wraith, MD. Graphic 75625 Version 3. 0 Sella spot grafide sella tursikada genişleme (Dysostosis multiplex ) Courtesy of Ed Wraith, MD. Graphic 75624 Version 3. 0
17 yaşında erkek hasta; Hurler-Scheie sendromu tanılı olguda korneal bulutlanma. Courtesy of Emil Kakkis, MD, Ph. D. Graphic 52924 Version 2. 0
İdrar mukopolisakkarid ve oligosakkarid atılımı (Willink Laboratuarı, Manchester) • • Oligosakkarid: Trisakkarid bandı izlendi. Siyalik asit: Normal Mukopolisakkarid: Normal YORUMU: Oligosakkarid paterni alfamannosidozu desteklemekte olup, enzim analizi önerildi. • Alfa-mannosidaz enzimi: 2. 6 umol/g/h (100 -800)
Alfa-mannosidoz • Lizozomal alfa-mannosidaz enziminin (MAN 2 B 1 mutasyonu, 19 p 13. 2 -q 12) eksikliği ile karakterize lizozomal bir depo hastalığı. • Alfa-mannosidaz enziminin eksikliğinde, lizozomlarda sindirilmemiş oligosakkarid molekülleri birçok sistemde birikir. • Görülme sıklığı: 1/500000 • OR kalıtım • Hurler-like sendrom (Öckerman, 1967) • Ayırıcı tanıda MPS başta olmak üzere diğer lizozomal depo hastalıkları düşünülmeli.
Oligosakkaridoz/Glikoproteinler
Klinik Özellikler • Fasyal dismorfizm: Hurler fenotipi (kaba yüz görünümü, makroglossi, belirgin alın, prognatizm) • Oküler bulgular: Şaşılık, korneal opasite, kırma kusuru • İşitme kaybı (non-progresif): İleti tipi ve/veya sensörinöral tipte • Hepatosplenomegali • Motor fonksiyonlarda bozukluk: Ataksi, ince motor becerilerde gerilik, kas güçsüzlüğü • Bilişsel gerilik • Psikiyatrik bulgular: Anksiyete bozukluğu, depresyon, psikoz • İmmun yetmezlik (hücresel ve humoral) • İskelet tutulumu (dizostozis multipleks, kifoskolyoz)
Alfa-mannosidoz • 3 klinik tipi mevcut. Ø Tip 1 (hafif): Genellikle 10 yaşından sonra tanı alır. Miyopati gözlenir, yavaş seyirlidir, iskelet anomalileri beklenmez. Ø Tip 2 (orta): Genellikle 10 yaşından önce tanı alır. İskelet anomalileri ve miyopati gözlenir ve yavaş seyir gösterir. Ø Tip 3 (ağır): Hızlı progresyon gösterir. Primer olarak santral sinir sistemi tutulumu ve tekrarlayan enfeksiyonlar nedeni ile erken yaşta kaybedilir.
Alfa-mannosidoz • Tanı: qİdrarda mannozdan zengin oligosakkarid atılımı (tek başına diagnostik değil) q. Kemik iliği incelemesinde ve periferik yaymada lenfositlerde vakuollerin varlığı (şüphe halinde tarama testi olarak kullanılabilir) q. Lökosit veya fibroblastlarda alfa-mannosidaz enzim düzeyi ölçümü q. Mutasyon analizi tayini (MAN 2 B 1 mutasyonu)
Alfa-mannosidoz • Tedavi: qÖzel eğitim ve rehabilitasyon desteği q. Baş çevresi takibi (hidrosefali!) q. Ortopedik cerrahi girişimler (komplikasyonlara yönelik) q. Tekrarlayan otit ve işitme kaybı açısından izlem q. KİT (hayatın ilk 10 yılı içinde, komplikasyonlar gelişmeden önce bir seçenek olabilir)
İzlem • Olgumuz halen özel eğitim desteği almakta, 3 -4 kelimesi mevcut, ancak cümle kuramıyor. • Şaşılık nedeni ile gözlük kullanıyor, işitme kaybı nedeni ile ventilasyon tüpü takılması önerildi. • Enzim tedavisi? • Hasta İ. Ü. İstanbul Tıp Fakültesi Beslenme ve Metabolizma polikliniğine yönlendirildi. • Ç. Nöroloji, Ç. Kardiyoloji, KBB, Göz ve Çocuk Ruh Sağlığı ve Hastalıkları kliniklerinde izlemine devam edilecek.
İzlem • Olguya enzimatik tanı konulduğunda annenin 3 haftalık gebe olduğu öğrenildi. • Aile prenatal tanı (MAN 2 B 1) açısından bilgilendirildi.
Özetle • Birçok sistemin birlikte tutulumu söz konusu olduğunda; o kaba yüz görünümü o hepatosplenomegali Lizozomal depo o motor ve bilişsel gerilik hastalıklarını o oküler bulgular (özellikle MPS) düşün! o işitme kaybı o İskelet sistemi tutulumu o Kardiyak tutulum
A , B y C. Pottery sample from the Tumaco-Tolita culture representing a possible case of Maroteaux Lamy syndrome. Mucopolysaccharidosis type VI (Maroteaux-Lamy syndrome) in the pre-Columbian culture of Colombia. Pachajoa H, Rodriguez CA 2014 Jun 30; 45(2): 85 -7.